
14.3: Medicamentos direcionados a outros microrganismos
- Page ID
- 181743

Objetivos de
- Descreva os mecanismos de ação associados a drogas que inibem a biossíntese da parede celular, a síntese de proteínas, a função da membrana, a síntese de ácidos nucléicos e a via metabólica.
Uma qualidade importante de um medicamento antimicrobiano é a toxicidade seletiva, o que significa que ele mata ou inibe seletivamente o crescimento de alvos microbianos, causando o mínimo ou nenhum dano ao hospedeiro. A maioria dos antimicrobianos atualmente em uso clínico são antibacterianos porque a célula procariótica fornece uma maior variedade de alvos exclusivos para toxicidade seletiva, em comparação com fungos, parasitas e vírus. Cada classe de medicamentos antibacterianos tem um modo de ação único (a maneira pela qual um medicamento afeta os micróbios no nível celular), e eles estão resumidos na Figura\(\PageIndex{1}\) e na Tabela\(\PageIndex{1}\).
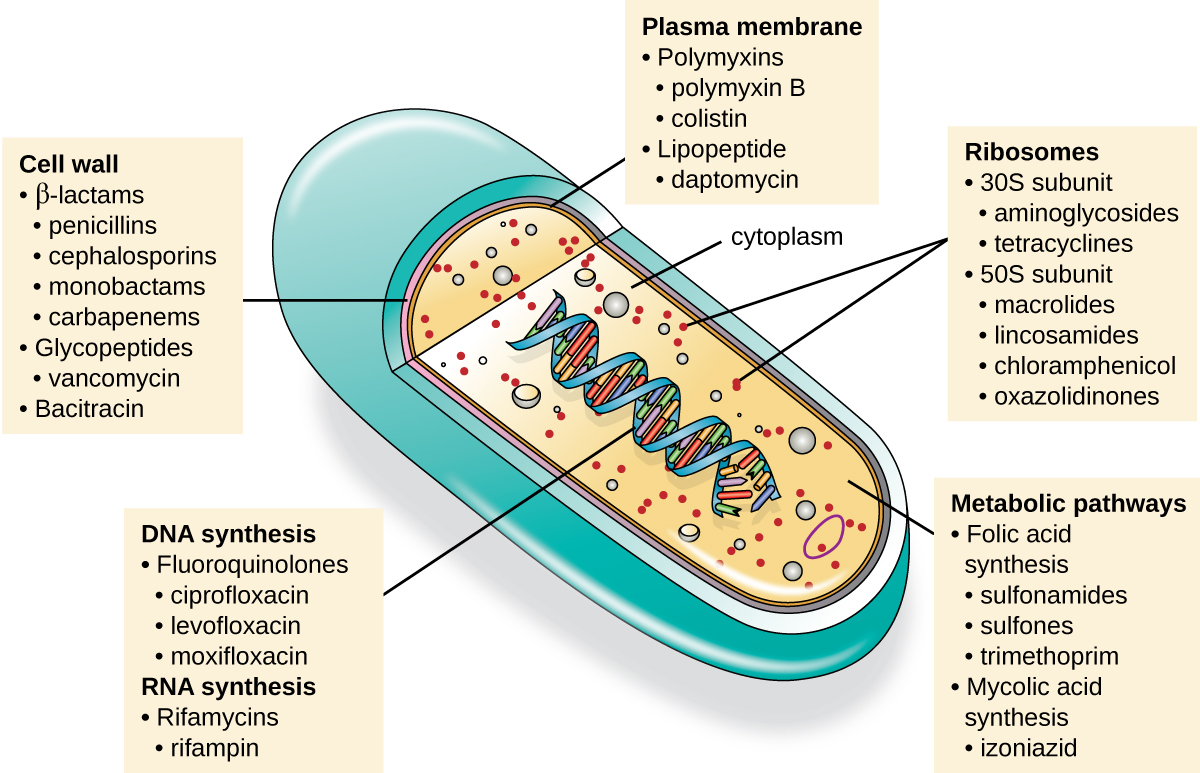
| Modo de ação | Alvo | Classe de drogas |
|---|---|---|
| Inibe a biossíntese da parede celular | Proteínas de ligação à penicilina | β-lactamas: penicilinas, cefalosporinas, monobactamas, carbapenêmicos |
| Subunidades de peptidoglicanos | Glicopeptídeos | |
| Transporte de subunidades de peptidoglicanos | Bacitracina | |
| Inibir a biossíntese de proteínas | Subunidade ribossômica 30S | Aminoglicosídeos, tetraciclinas |
| Subunidade ribossômica 50S | Macrolídeos, lincosamidas, cloranfenicol, oxazolidinonas | |
| Romper as membranas | Lipopolissacarídeo, membranas interna e externa | Polimixina B, colistina, daptomicina |
| Inibir a síntese de ácido nucleico | RNA | Rifamicina |
| DNA | Fluoroquinolonas | |
| Antimetabólitos | Enzima de síntese de ácido fólico | Sulfonamidas, trimetoprim |
| Enzima de síntese de ácido micólico | Hidrazida do ácido isonicotínico | |
| Inibidor da adenosina trifosfato micobacteriano (ATP) sintase | ATP sintase micobacteriana | Diarilquinolina |
Inibidores da biossíntese da parede celular
Várias classes diferentes de antibacterianos bloqueiam as etapas da biossíntese do peptidoglicano, tornando as células mais suscetíveis à lise osmótica (Tabela\(\PageIndex{2}\)). Portanto, os antibacterianos que têm como alvo a biossíntese da parede celular são bactericidas em sua ação. Como as células humanas não produzem peptidoglicano, esse modo de ação é um excelente exemplo de toxicidade seletiva.
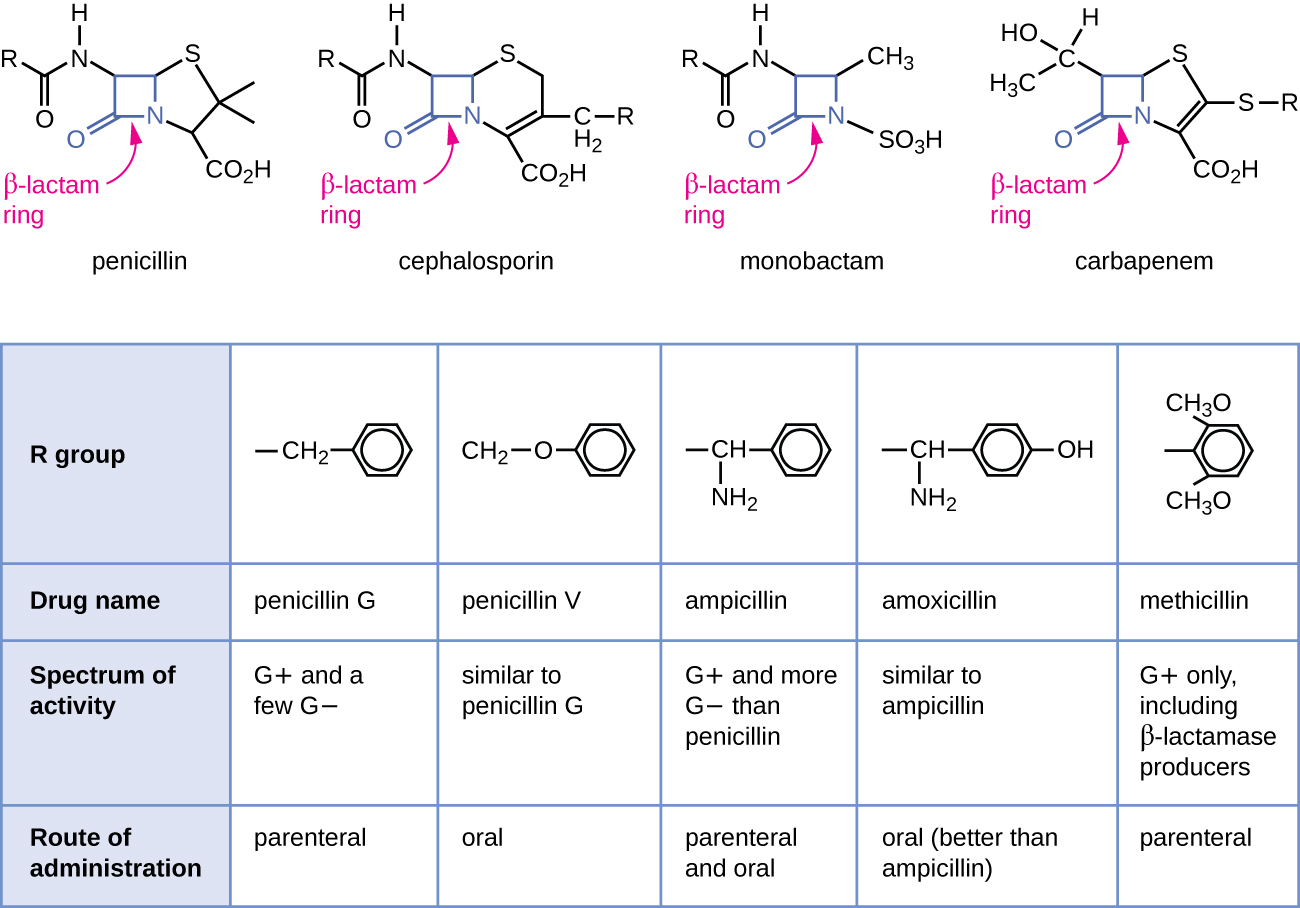
A penicilina, o primeiro antibiótico descoberto, é um dos vários antibacterianos dentro de uma classe chamada β-lactamas. Esse grupo de compostos inclui as penicilinas, cefalosporinas, monobactâmicos e carbapenêmicos e é caracterizado pela presença de um anel β-lactâmico encontrado na estrutura central da molécula do fármaco (Figura\(\PageIndex{2}\)). Os antibacterianos β-lactâmicos bloqueiam a reticulação das cadeias peptídicas durante a biossíntese de novos peptidoglicanos na parede celular bacteriana. Eles são capazes de bloquear esse processo porque a estrutura β-lactâmica é semelhante à estrutura do componente da subunidade do peptidoglicano que é reconhecida pela enzima transpeptidase de reticulação, também conhecida como proteína de ligação à penicilina (PBP). Embora o anel β-lactâmico deva permanecer inalterado para que esses medicamentos mantenham sua atividade antibacteriana, mudanças químicas estratégicas nos grupos R permitiram o desenvolvimento de uma ampla variedade de medicamentos β-lactâmicos semissintéticos com maior potência, espectro expandido de atividade e meia-vida mais longa para melhor dosagem, entre outras características.
A penicilina G e a penicilina V são antibióticos naturais de fungos e são principalmente ativos contra patógenos bacterianos gram-positivos e alguns patógenos bacterianos gram-negativos, como a Pasteurella multocida. A figura\(\PageIndex{2}\) resume o desenvolvimento semissintético de algumas das penicilinas. A adição de um grupo amino (-NH 2) à penicilina G criou as aminopenicilinas (ou seja, ampicilina e amoxicilina) que aumentaram o espectro de atividade contra mais patógenos gram-negativos. Além disso, a adição de um grupo hidroxila (-OH) à amoxicilina aumentou a estabilidade ácida, o que permite uma melhor absorção oral. A meticilina é uma penicilina semissintética que foi desenvolvida para combater a disseminação de enzimas (penicilinases) que estavam inativando as outras penicilinas. A mudança do grupo R da penicilina G para o grupo dimetoxifenil mais volumoso protegeu o anel β-lactâmico da destruição enzimática pelas penicilinases, resultando na primeira penicilina resistente à penicilinase.
Semelhante às penicilinas, as cefalosporinas contêm um anel β-lactâmico (Figura\(\PageIndex{2}\)) e bloqueiam a atividade da transpeptidase das proteínas de ligação à penicilina. No entanto, o anel β-lactâmico das cefalosporinas é fundido a um anel de seis membros, em vez do anel de cinco membros encontrado nas penicilinas. Essa diferença química fornece às cefalosporinas uma maior resistência à inativação enzimática por β-lactamases. O medicamento cefalosporina C foi originalmente isolado do fungo Cephalosporium acremonium na década de 1950 e tem um espectro de atividade semelhante ao da penicilina contra bactérias gram-positivas, mas é ativo contra mais bactérias gram-negativas do que a penicilina. Outra diferença estrutural importante é que a cefalosporina C possui dois grupos R, em comparação com apenas um grupo R para a penicilina, o que proporciona maior diversidade nas alterações químicas e no desenvolvimento de cefalosporinas semissintéticas. A família das cefalosporinas semissintéticas é muito maior do que as penicilinas, e essas drogas foram classificadas em gerações com base principalmente em seu espectro de atividade, aumentando no espectro das cefalosporinas de primeira geração de espectro estreito para as de amplo espectro, de quarta geração cefalosporinas. Foi desenvolvida uma nova cefalosporina de quinta geração que é ativa contra o Staphylococcus aureus resistente à meticilina (MRSA).
Os carbapenêmicos e monobactâmicos também têm um anel β-lactâmico como parte de sua estrutura central e inibem a atividade da transpeptidase das proteínas de ligação à penicilina. O único monobactama usado clinicamente é o aztreonam. É um antibacteriano de espectro estreito com atividade apenas contra bactérias gram-negativas. Em contraste, a família dos carbapenêmicos inclui uma variedade de medicamentos semissintéticos (imipenem, meropenem e doripenem) que fornecem atividade de amplo espectro contra patógenos bacterianos gram-positivos e gram-negativos.
O medicamento vancomicina, membro de uma classe de compostos chamados glicopeptídeos, foi descoberto na década de 1950 como um antibiótico natural do actinomiceto Amycolatopsis orientalis. Semelhante aos β-lactâmicos, a vancomicina inibe a biossíntese da parede celular e é bactericida. No entanto, em contraste com os β-lactâmicos, a estrutura da vancomicina não é semelhante à das subunidades do peptidoglicano da parede celular e não inativa diretamente as proteínas de ligação à penicilina. Pelo contrário, a vancomicina é uma molécula muito grande e complexa que se liga ao final da cadeia peptídica dos precursores da parede celular, criando um bloqueio estrutural que impede que as subunidades da parede celular sejam incorporadas à crescente espinha dorsal da N-acetilglucosamina e do ácido N-acetilmurâmico (NAM-NAG) do peptidoglicano estrutura (transglicosilação). A vancomicina também bloqueia estruturalmente a transpeptidação. A vancomicina é bactericida contra patógenos bacterianos gram-positivos, mas não é ativa contra bactérias gram-negativas devido à sua incapacidade de penetrar na membrana externa protetora.
A droga bacitracina consiste em um grupo de antibióticos peptídicos estruturalmente similares originalmente isolados de Bacillus subtilis. A bacitracina bloqueia a atividade de uma molécula específica da membrana celular responsável pelo movimento dos precursores do peptidoglicano do citoplasma para o exterior da célula, impedindo sua incorporação na parede celular. A bacitracina é eficaz contra uma ampla gama de bactérias, incluindo organismos gram-positivos encontrados na pele, como Staphylococcus e Streptococcus. Embora possa ser administrada por via oral ou intramuscular em algumas circunstâncias, demonstrou-se que a bacitracina é nefrotóxica (danifica os rins). Portanto, é mais comumente combinado com neomicina e polimixina em pomadas tópicas, como a neosporina.
| Mecanismo de ação | Classe de drogas | Medicamentos específicos | Natural ou semissintético | Espectro de atividade |
|---|---|---|---|---|
| Interaja diretamente com os PBPs e iniba a atividade da transpeptidase | Penicilinas | Penicilina G, penicilina V | Natural | Espectro estreito contra bactérias gram-positivas e algumas gram-negativas |
| Ampicilina, amoxicilina | Semissintético | Espectro estreito contra bactérias gram-positivas, mas com espectro gram-negativo aumentado | ||
| Meticilina | Semissintético | Espectro estreito somente contra bactérias gram-positivas, incluindo cepas produtoras de penicilinase | ||
| Cefalosporinas | Cefalosporina C | Natural | Espectro estreito semelhante à penicilina, mas com aumento do espectro gram-negativo | |
| Cefalosporinas de primeira geração | Semissintético | Espectro estreito semelhante à cefalosporina C | ||
| Cefalosporinas de segunda geração | Semissintético | Espectro estreito, mas com maior espectro gram-negativo em comparação com a primeira geração | ||
| Cefalosporinas de terceira e quarta geração | Semissintético | Amplo espectro contra bactérias gram-positivas e gram-negativas, incluindo alguns produtores de β-lactamase | ||
| Cefalosporinas de quinta geração | Semissintético | Amplo espectro contra bactérias gram-positivas e gram-negativas, incluindo MRSA | ||
| Monobactâmicos | Aztreonam | Semissintético | Espectro estreito contra bactérias gram-negativas, incluindo alguns produtores de β-lactamase | |
| Carbapenêmicos | Imipenem, meropenem, doripenem | Semissintético | O mais amplo espectro de β-lactamas contra bactérias gram-positivas e gram-negativas, incluindo muitos produtores de β-lactamase | |
| Moléculas grandes que se ligam à cadeia peptídica das subunidades do peptidoglicano, bloqueando a transglicosilação e a transpeptidação | Glicopeptídeos | Vancomicina | Natural | Espectro estreito somente contra bactérias gram-positivas, incluindo cepas multirresistentes |
| Bloquear o transporte de subunidades de peptidoglicanos através da membrana citoplasmática | Bacitracina | Bacitracina | Natural | Amplo espectro contra bactérias gram-positivas e gram-negativas |
Exercício\(\PageIndex{1}\)
Descreva o modo de ação dos β-lactâmicos.
Inibidores da biossíntese de proteínas
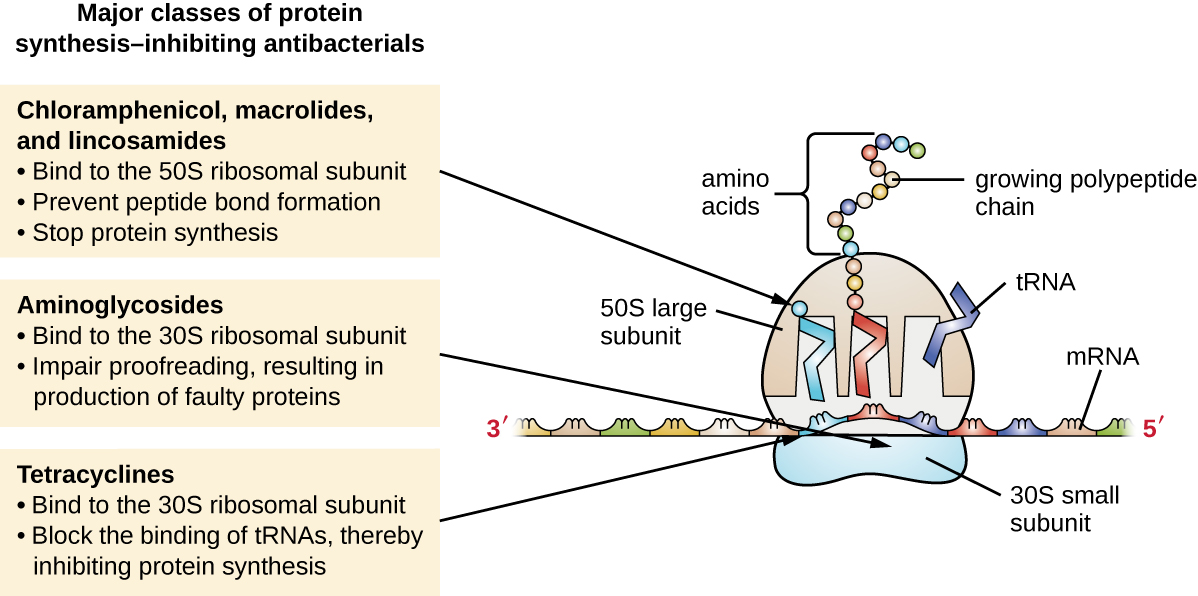
Os ribossomos citoplasmáticos encontrados em células animais (80S) são estruturalmente distintos daqueles encontrados em células bacterianas (70S), tornando a biossíntese de proteínas um bom alvo seletivo para medicamentos antibacterianos. Vários tipos de inibidores da biossíntese de proteínas são discutidos nesta seção e estão resumidos na Figura\(\PageIndex{3}\).
Inibidores da síntese de proteínas que se ligam à subunidade 30S
Os aminoglicosídeos são medicamentos antibacterianos grandes e altamente polares que se ligam à subunidade 30S dos ribossomos bacterianos, prejudicando a capacidade de revisão do complexo ribossômico. Esse comprometimento causa incompatibilidade entre códons e anticódons, resultando na produção de proteínas com aminoácidos incorretos e proteínas encurtadas que se inserem na membrana citoplasmática. A ruptura da membrana citoplasmática pelas proteínas defeituosas mata as células bacterianas. Os aminoglicosídeos, que incluem medicamentos como estreptomicina, gentamicina, neomicina e canamicina, são potentes antibacterianos de amplo espectro. No entanto, foi demonstrado que os aminoglicosídeos são nefrotóxicos (danificam os rins), neurotóxicos (danificam o sistema nervoso) e ototóxicos (danificam o ouvido).
Outra classe de compostos antibacterianos que se ligam à subunidade 30S são as tetraciclinas. Em contraste com os aminoglicosídeos, esses medicamentos são bacteriostáticos e inibem a síntese protéica ao bloquear a associação de tRNAs com o ribossomo durante a tradução. As tetraciclinas de ocorrência natural produzidas por várias cepas de Streptomyces foram descobertas pela primeira vez na década de 1940, e várias tetraciclinas semissintéticas, incluindo doxiciclina e tigeciclina, também foram produzidas. Embora as tetraciclinas tenham amplo espectro em sua cobertura de patógenos bacterianos, os efeitos colaterais que podem limitar seu uso incluem fototoxicidade, descoloração permanente dos dentes em desenvolvimento e toxicidade hepática com altas doses ou em pacientes com insuficiência renal.
Inibidores da síntese de proteínas que se ligam à subunidade 50S
Existem várias classes de medicamentos antibacterianos que atuam por meio da ligação à subunidade 50S dos ribossomos bacterianos. Os medicamentos antibacterianos macrolídeos têm uma estrutura em anel grande e complexa e fazem parte de uma classe maior de metabólitos secundários produzidos naturalmente chamados policetídeos, compostos complexos produzidos de forma gradual por meio da adição repetida de unidades de dois carbonos por um mecanismo semelhante ao usado para gorduras síntese ácida. Os macrolídeos são fármacos bacteriostáticos de amplo espectro que bloqueiam o alongamento das proteínas ao inibir a formação de ligações peptídicas entre combinações específicas de aminoácidos. O primeiro macrolídeo foi a eritromicina. Foi isolado em 1952 do Streptomyces erythreus e evita a translocação. Os macrolídeos semissintéticos incluem azitromicina e telitromicina. Em comparação com a eritromicina, a azitromicina tem um espectro de atividade mais amplo, menos efeitos colaterais e uma meia-vida significativamente mais longa (1,5 horas para eritromicina versus 68 horas para azitromicina) que permite a dosagem uma vez ao dia e um curto curso de terapia de 3 dias (ou seja, formulação de Zpac) para a maioria das infecções. A telitromicina é o primeiro semissintético da classe conhecida como cetólidos. Embora a telitromicina mostre maior potência e atividade contra patógenos resistentes a macrolídeos, a Food and Drug Administration (FDA) dos EUA limitou seu uso ao tratamento da pneumonia adquirida na comunidade e exige o rótulo de “alerta de caixa preta” mais forte para o medicamento devido à grave hepatotoxicidade.
As lincosamidas incluem a lincomicina produzida naturalmente e a clindamicina semissintética. Embora estruturalmente distintas dos macrolídeos, as lincosamidas são semelhantes em seu modo de ação aos macrolídeos por meio da ligação à subunidade ribossômica 50S e impedindo a formação de ligações peptídicas. As lincosamidas são particularmente ativas contra infecções estreptocócicas e estafilocócicas.
O fármaco cloranfenicol representa mais uma classe estruturalmente distinta de antibacterianos que também se ligam ao ribossomo 50S, inibindo a formação de ligações peptídicas. O cloranfenicol, produzido por Streptomyces venezuelae, foi descoberto em 1947; em 1949, tornou-se o primeiro antibiótico de amplo espectro aprovado pelo FDA. Embora seja um antibiótico natural, também é facilmente sintetizado e foi o primeiro medicamento antibacteriano produzido sinteticamente em massa. Como resultado de sua produção em massa, cobertura de amplo espectro e capacidade de penetrar nos tecidos com eficiência, o cloranfenicol foi historicamente usado para tratar uma ampla gama de infecções, desde meningite até febre tifóide e conjuntivite. Infelizmente, efeitos colaterais graves, como a síndrome do bebê cinzento letal e a supressão da produção de medula óssea, limitaram seu papel clínico. O cloranfenicol também causa anemia de duas maneiras diferentes. Um mecanismo envolve o direcionamento dos ribossomos mitocondriais nas células-tronco hematopoiéticas, causando uma supressão reversível e dependente da dose da produção de células sanguíneas. Quando a dosagem de cloranfenicol é interrompida, a produção de células sanguíneas volta ao normal. Esse mecanismo destaca a semelhança entre os ribossomos 70S de bactérias e os ribossomos 70S em nossas mitocôndrias. O segundo mecanismo da anemia é idiossincrático (ou seja, o mecanismo não é compreendido) e envolve uma perda letal irreversível da produção de células sanguíneas conhecida como anemia aplástica. Esse mecanismo de anemia aplástica não depende da dose e pode se desenvolver após a interrupção da terapia. Devido a questões de toxicidade, o uso de cloranfenicol em humanos agora é raro nos Estados Unidos e está limitado a infecções graves que não podem ser tratadas com antibióticos menos tóxicos. Como seus efeitos colaterais são muito menos graves em animais, ele é usado em medicina veterinária.
As oxazolidinonas, incluindo a linezolida, são uma nova classe de inibidores de síntese de proteínas sintéticas de amplo espectro que se ligam à subunidade ribossômica 50S de bactérias gram-positivas e gram-negativas. No entanto, seu mecanismo de ação parece um pouco diferente do dos outros inibidores da síntese de proteínas de ligação à subunidade 50S já discutidos. Em vez disso, eles parecem interferir na formação do complexo de iniciação (associação da subunidade 50S, subunidade 30S e outros fatores) para tradução e impedem a translocação da proteína em crescimento do sítio A ribossômico para o sítio P. A tabela\(\PageIndex{3}\) resume os inibidores da síntese de proteínas.
| Alvo molecular | Mecanismo de ação | Classe de drogas | Medicamentos específicos | Bacteriostático ou bactericida | Espectro de atividade |
|---|---|---|---|---|---|
| Subunidade 30S | Causa incompatibilidade entre códons e anticódons, levando a proteínas defeituosas que se inserem e interrompem a membrana citoplasmática | Aminoglicosídeos | Estreptomicina, gentamicina, neomicina, canamicina | Bactericida | Amplo espectro |
| Bloqueia a associação de tRNAs com ribossomo | Tetraciclinas | Tetraciclina, doxiciclina, tigeciclina | Bacteriostático | Amplo espectro | |
| Subunidade 50S | Bloqueia a formação de ligações peptídicas entre aminoácidos | Macrolídeos | Eritromicina, azitromicina, telitromicina | Bacteriostático | Amplo espectro |
| Lincosamidas | Lincomicina, clindamicina | Bacteriostático | espectro estreito | ||
| Não aplicável | Cloranfenicol | Bacteriostático | Amplo espectro | ||
| Interfere na formação do complexo de iniciação entre as subunidades 50S e 30S e outros fatores. | Oxazolidinonas | Linezolida | Bacteriostático | Amplo espectro |
Exercício\(\PageIndex{2}\)
Compare e contraste os diferentes tipos de inibidores da síntese de proteínas.
Inibidores da função da membrana
Um pequeno grupo de antibacterianos tem como alvo a membrana bacteriana como seu modo de ação (Tabela\(\PageIndex{4}\)). As polimixinas são antibióticos polipeptídicos naturais que foram descobertos pela primeira vez em 1947 como produtos do Bacillus polymyxa; somente a polimixina B e a polimixina E (colistina) foram usadas clinicamente. Eles são lipofílicos com propriedades semelhantes a detergentes e interagem com o componente lipopolissacarídeo da membrana externa das bactérias gram-negativas, acabando por romper as membranas externa e interna e matando as células bacterianas. Infelizmente, o mecanismo de direcionamento por membrana não é uma toxicidade seletiva, e esses medicamentos também têm como alvo e danificam a membrana das células do rim e do sistema nervoso quando administrados sistemicamente. Devido a esses efeitos colaterais graves e sua baixa absorção pelo trato digestivo, a polimixina B é usada em pomadas antibióticas tópicas de venda livre (por exemplo, Neosporina), e a colistina oral foi historicamente usada apenas para descontaminação intestinal para prevenir infecções originadas por micróbios intestinais em pacientes imunocomprometidos ou para aqueles submetidos a determinadas cirurgias abdominais. No entanto, o surgimento e a disseminação de patógenos multirresistentes levaram ao aumento do uso de colistina intravenosa em hospitais, geralmente como um medicamento de último recurso para tratar infecções graves. A daptomicina antibacteriana é um lipopeptídeo cíclico produzido pelo Streptomyces roseosporus que parece funcionar como as polimixinas, inserindo-se na membrana celular bacteriana e rompendo-a. No entanto, em contraste com a polimixina B e a colistina, que têm como alvo apenas bactérias gram-negativas, a daptomicina tem como alvo específico as bactérias gram-positivas. Geralmente é administrado por via intravenosa e parece ser bem tolerado, mostrando toxicidade reversível nos músculos esqueléticos.
| Mecanismo de ação | Classe de drogas | Medicamentos específicos | Espectro de atividade | Uso clínico |
|---|---|---|---|---|
| Interage com o lipopolissacarídeo na membrana externa das bactérias gram-negativas, matando a célula por meio da eventual ruptura da membrana externa e da membrana citoplasmática | Polimixinas | Polimixina B | Espectro estreito contra bactérias gram-negativas, incluindo cepas multirresistentes | Preparações tópicas para prevenir infecções em feridas |
| Polimixina E (colistina) | Espectro estreito contra bactérias gram-negativas, incluindo cepas multirresistentes | Dosagem oral para descontaminar intestinos para prevenir infecções em pacientes imunocomprometidos ou pacientes submetidos a cirurgias/procedimentos invasivos. | ||
| Dosagem intravenosa para tratar infecções sistêmicas graves causadas por patógenos multirresistentes | ||||
| Insere-se na membrana citoplasmática de bactérias gram-positivas, rompendo a membrana e matando a célula | Lipopeptídeo | Daptomicina | Espectro estreito contra bactérias gram-positivas, incluindo cepas multirresistentes | Infecções complicadas da pele e da estrutura da pele e bacteremia causadas por patógenos gram-positivos, incluindo MRSA |
Exercício\(\PageIndex{3}\)
Como as polimixinas inibem a função da membrana?
Inibidores da síntese de ácidos nucleicos
Alguns medicamentos antibacterianos atuam inibindo a síntese de ácidos nucléicos (Tabela\(\PageIndex{5}\)). Por exemplo, o metronidazol é um membro semissintético da família dos nitroimidazóis que também é um antiprotozoário. Ele interfere na replicação do DNA nas células-alvo. A droga rifampicina é um membro semissintético da família das rifamicinas e funciona bloqueando a atividade da RNA polimerase em bactérias. As enzimas da RNA polimerase nas bactérias são estruturalmente diferentes das dos eucariotos, proporcionando toxicidade seletiva contra células bacterianas. É usado para o tratamento de uma variedade de infecções, mas seu uso principal, geralmente em um coquetel com outros medicamentos antibacterianos, é contra micobactérias causadoras da tuberculose. Apesar da seletividade de seu mecanismo, a rifampicina pode induzir enzimas hepáticas para aumentar o metabolismo de outras drogas administradas (antagonismo), levando à hepatotoxicidade (toxicidade hepática) e influenciando negativamente a biodisponibilidade e o efeito terapêutico dos medicamentos associados.
Um membro da família das quinolonas, um grupo de antimicrobianos sintéticos, é o ácido nalidíxico. Foi descoberto em 1962 como um subproduto durante a síntese da cloroquina, um medicamento antimalárico. O ácido nalidíxico inibe seletivamente a atividade da DNA girase bacteriana, bloqueando a replicação do DNA. Modificações químicas na estrutura original da quinolona resultaram na produção de fluoroquinolonas, como ciprofloxacina e levofloxacina, que também inibem a atividade da DNA girase. A ciprofloxacina e a levofloxacina são eficazes contra um amplo espectro de bactérias gram-positivas ou gram-negativas e estão entre os antibióticos mais comumente prescritos usados para tratar uma ampla gama de infecções, incluindo infecções do trato urinário, infecções respiratórias, infecções abdominais e infecções de pele. No entanto, apesar de sua toxicidade seletiva contra a DNA girase, os efeitos colaterais associados a diferentes fluoroquinolonas incluem fototoxicidade, neurotoxicidade, cardiotoxicidade, disfunção do metabolismo da glicose e aumento do risco de ruptura do tendão.
| Mecanismos de ação | Classe de drogas | Medicamentos específicos | Espectro de atividade | Uso clínico |
|---|---|---|---|---|
| Inibe a atividade da RNA polimerase bacteriana e bloqueia a transcrição, matando a célula | Rifamicina | Rifampicina | Espectro estreito com atividade contra bactérias gram-positivas e limitadas de bactérias gram-negativas. Também ativo contra o Mycobacterium tuberculosis. | Terapia combinada para tratamento da tuberculose |
| Inibe a atividade da DNA girase e bloqueia a replicação do DNA, matando a célula | Fluoroquinolonas | Ciprofloxacina, ofloxacina, moxifloxacina | Amplo espectro contra bactérias gram-positivas e gram-negativas | Grande variedade de infecções cutâneas e sistêmicas |
Exercício\(\PageIndex{4}\)
Por que os inibidores da síntese de ácidos nucleicos bacterianos não atingem as células hospedeiras?
Inibidores das vias metabólicas
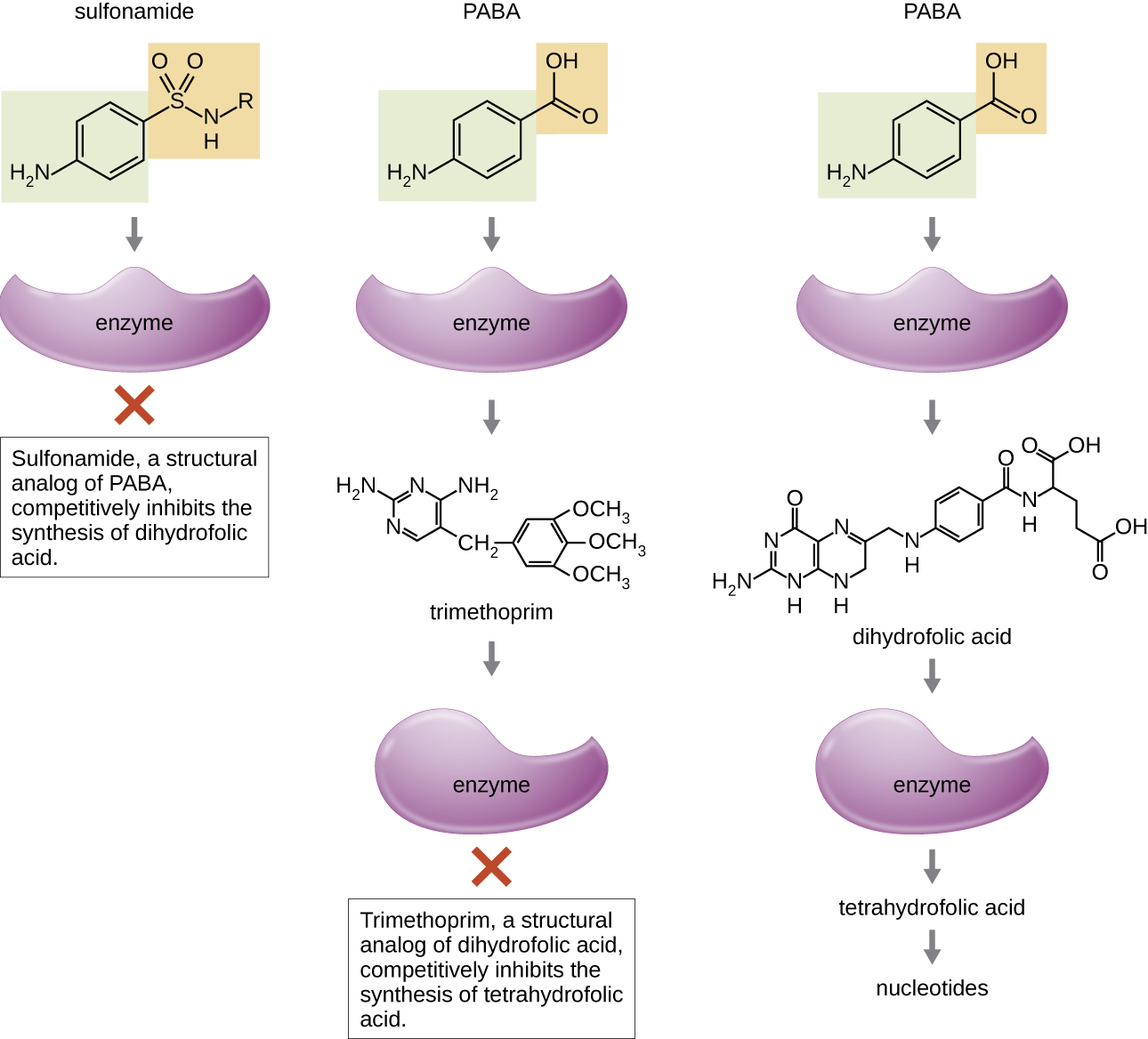
Algumas drogas sintéticas controlam infecções bacterianas funcionando como antimetabólitos, inibidores competitivos de enzimas metabólicas bacterianas (Tabela\(\PageIndex{6}\)). As sulfonamidas (sulfa-fármacos) são os agentes antibacterianos sintéticos mais antigos e são análogos estruturais do ácido para-aminobenzóico (PABA), um intermediário precoce na síntese do ácido fólico (Figura\(\PageIndex{4}\)). Ao inibir a enzima envolvida na produção de ácido dihidrofólico, as sulfonamidas bloqueiam a biossíntese bacteriana do ácido fólico e, posteriormente, das pirimidinas e purinas necessárias para a síntese do ácido nucleico. Esse mecanismo de ação fornece inibição bacteriostática do crescimento contra um amplo espectro de patógenos gram-positivos e gram-negativos. Como os humanos obtêm ácido fólico dos alimentos em vez de sintetizá-lo intracelularmente, as sulfonamidas são seletivamente tóxicas para as bactérias. No entanto, reações alérgicas aos medicamentos com sulfa são comuns. As sulfonas são estruturalmente semelhantes às sulfonamidas, mas não são comumente usadas hoje, exceto para o tratamento da doença de Hansen (hanseníase).
O trimetoprim é um composto antimicrobiano sintético que serve como antimetabólito na mesma via de síntese do ácido fólico que as sulfonamidas. No entanto, o trimetoprim é um análogo estrutural do ácido dihidrofólico e inibe uma etapa posterior na via metabólica (Figura\(\PageIndex{4}\)). O trimetoprim é usado em combinação com o sulfa-fármaco sulfametoxazol para tratar infecções do trato urinário, infecções de ouvido e bronquite. Conforme discutido, a combinação de trimetoprim e sulfametoxazol é um exemplo de sinergia antibacteriana. Quando usado sozinho, cada antimetabólito só diminui a produção de ácido fólico até um nível em que ocorre a inibição bacteriostática do crescimento. No entanto, quando usada em combinação, a inibição de ambas as etapas da via metabólica diminui a síntese de ácido fólico a um nível letal para a célula bacteriana. Devido à importância do ácido fólico durante o desenvolvimento fetal, o uso de sulfa e trimetoprim deve ser cuidadosamente considerado durante o início da gravidez.
A droga isoniazida é um antimetabólito com toxicidade específica para micobactérias e há muito tempo é usada em combinação com rifampicina ou estreptomicina no tratamento da tuberculose. É administrado como um pró-fármaco, requerendo ativação através da ação de uma enzima peroxidase bacteriana intracelular, formando isoniazida-nicotinamida adenina dinucleotídeo (NAD) e isoniazida-nicotinamida adenina dinucleotídeo fosfato (NADP), impedindo a síntese de ácido micólico, que é essencial para paredes celulares de micobactérias. Os possíveis efeitos colaterais do uso de isoniazida incluem hepatotoxicidade, neurotoxicidade e toxicidade hematológica (anemia).
| Alvo da via metabólica | Mecanismo de ação | Classe de drogas | Medicamentos específicos | Espectro de atividade |
|---|---|---|---|---|
| Síntese de ácido fólico | Inibe a enzima envolvida na produção de ácido dihidrofólico | Sulfonamidas | Sulfametoxazol | Amplo espectro contra bactérias gram-positivas e gram-negativas |
| Sulfonas | Dapsona | |||
| Inibe a enzima envolvida na produção de ácido tetrahidrofólico | Não aplicável | Trimetoprim | Amplo espectro contra bactérias gram-positivas e gram-negativas | |
| Síntese de ácido micólico | Interfere com a síntese do ácido micólico | Não aplicável | Isoniazida | Espectro estreito contra Mycobacterium spp., incluindo M. tuberculosis |
Exercício\(\PageIndex{5}\)
Como as sulfonamidas e o trimetoprim atacam seletivamente as bactérias?
Inibidor da ATP sintase
A bedaquilina, representando a classe antibacteriana sintética de compostos chamados diarilquinolonas, usa um novo modo de ação que inibe especificamente o crescimento de micobactérias. Embora o mecanismo específico ainda não tenha sido elucidado, esse composto parece interferir na função das ATP sintases, talvez interferindo no uso do gradiente de íons hidrogênio para síntese de ATP por fosforilação oxidativa, levando à redução da produção de ATP. Devido aos seus efeitos colaterais, incluindo hepatotoxicidade e arritmia cardíaca potencialmente letal, seu uso é reservado para casos graves de tuberculose, caso contrário não tratáveis.
Foco clínico: Parte 2
Ao ler minuciosamente o histórico de saúde de Marisa, o médico percebeu que durante sua hospitalização no Vietnã, ela foi cateterizada e recebeu os antimicrobianos ceftazidima e metronidazol. Ao saber disso, o médico solicitou uma tomografia computadorizada do abdômen de Marisa para descartar apendicite; o médico também solicitou exames de sangue para verificar se ela tinha uma contagem elevada de glóbulos brancos e solicitou um exame de urina e urocultura para verificar a presença de glóbulos brancos, glóbulos vermelhos e bactérias .
A amostra de urina de Marisa deu positivo para a presença de bactérias, indicando uma infecção do trato urinário (ITU). O médico prescreveu ciprofloxacina. Enquanto isso, sua urina foi cultivada para cultivar a bactéria para testes adicionais.
Exercício\(\PageIndex{6}\)
- Quais tipos de antimicrobianos são normalmente prescritos para ITUs?
- Com base nos medicamentos antimicrobianos que ela recebeu no Vietnã, qual dos antimicrobianos para o tratamento de uma ITU você previa ser ineficaz?
Conceitos principais e resumo
- Os compostos antibacterianos apresentam toxicidade seletiva, em grande parte devido às diferenças entre a estrutura celular procariótica e eucariótica.
- Os inibidores da síntese da parede celular, incluindo os β-lactâmicos, os glicopeptídeos e a bacitracina, interferem na síntese de peptidoglicanos, tornando as células bacterianas mais propensas à lise osmótica.
- Há uma variedade de inibidores da síntese de proteínas bacterianas de amplo espectro que visam seletivamente o ribossomo procariótico 70S, incluindo aqueles que se ligam à subunidade 30S (aminoglicosídeos e tetraciclinas) e outros que se ligam à subunidade 50S (macrolídeos, lincosamidas, cloranfenicol e oxazolidinonas).
- As polimixinas são antibióticos polipeptídicos lipofílicos que têm como alvo o componente lipopolissacarídeo das bactérias gram-negativas e, em última análise, interrompem a integridade das membranas externa e interna dessas bactérias.
- Os inibidores da síntese de ácido nucléico rifamicinas e fluoroquinolonas têm como alvo a transcrição do RNA bacteriano e a replicação do DNA, respectivamente.
- Alguns medicamentos antibacterianos são antimetabólitos, atuando como inibidores competitivos das enzimas metabólicas bacterianas. As sulfonamidas e o trimetoprim são antimetabólitos que interferem na síntese bacteriana do ácido fólico. A isoniazida é um antimetabólito que interfere na síntese do ácido micólico nas micobactérias.