
14.4: Considerações clínicas
- Page ID
- 181746

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Objetivos de
- Explicar as diferenças entre os modos de ação dos medicamentos que têm como alvo fungos, protozoários, helmintos e vírus
Como fungos, protozoários e helmintos são eucarióticos, suas células são muito semelhantes às células humanas, dificultando o desenvolvimento de medicamentos com toxicidade seletiva. Além disso, os vírus se replicam nas células hospedeiras humanas, dificultando o desenvolvimento de medicamentos seletivamente tóxicos para vírus ou células infectadas por vírus. Apesar desses desafios, existem medicamentos antimicrobianos que têm como alvo fungos, protozoários, helmintos e vírus, e alguns até têm como alvo mais de um tipo de micróbio. Tabela\(\PageIndex{1}\), Tabela\(\PageIndex{2}\)\(\PageIndex{3}\), Tabela e Tabela\(\PageIndex{4}\) fornecem exemplos de medicamentos antimicrobianos nessas várias classes.
Medicamentos antifúngicos
O modo de ação mais comum dos medicamentos antifúngicos é a ruptura da membrana celular. Os antifúngicos aproveitam as pequenas diferenças entre fungos e humanos nas vias bioquímicas que sintetizam esteróis. Os esteróis são importantes para manter a fluidez adequada da membrana e, portanto, o funcionamento adequado da membrana celular. Para a maioria dos fungos, o esterol de membrana predominante é o ergosterol. Como as membranas celulares humanas usam colesterol, em vez do ergosterol, os medicamentos antifúngicos que visam a síntese de ergosterol são seletivamente tóxicos (Figura\(\PageIndex{1}\)).
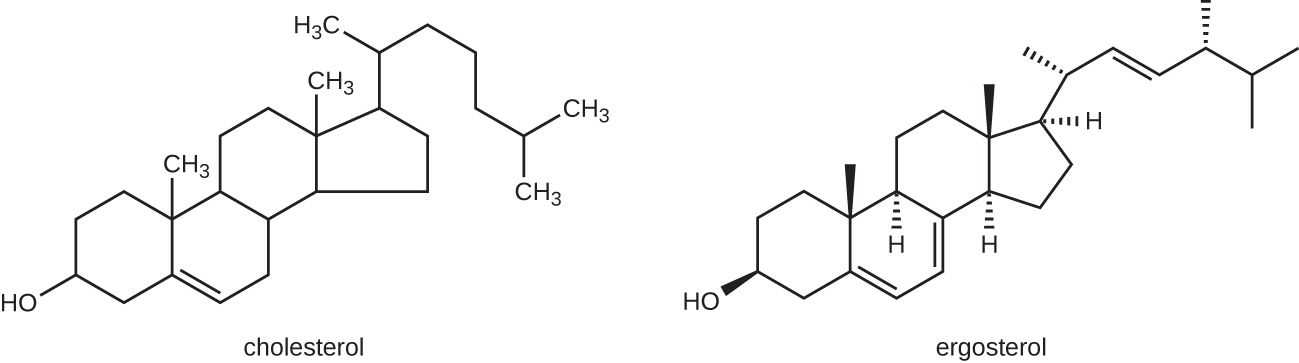
Os imidazóis são fungicidas sintéticos que interrompem a biossíntese do ergosterol; eles são comumente usados em aplicações médicas e também na agricultura para impedir que as sementes e as safras colhidas se moldem. Os exemplos incluem miconazol, cetoconazol e clotrimazol, que são usados para tratar infecções fúngicas da pele, como micose, especificamente tinea pedis (pé de atleta), tinea cruris (coceira de atleta) e tinea corporis. Essas infecções são comumente causadas por dermatófitos dos gêneros Trichophyton, Epidermophyton e Microsporum. O miconazol também é usado predominantemente para o tratamento de infecções vaginais por leveduras causadas pelo fungo Candida, e o cetoconazol é usado para o tratamento da tinea versicolor e da caspa, que podem ser causadas pelo fungo Malassezia.
Os medicamentos triazólicos, incluindo o fluconazol, também inibem a biossíntese do ergosterol. No entanto, eles podem ser administrados por via oral ou intravenosa para o tratamento de vários tipos de infecções fúngicas sistêmicas, incluindo candidíase oral e meningite criptocócica, ambas prevalentes em pacientes com AIDS. Os triazóis também apresentam toxicidade mais seletiva, em comparação com os imidazóis, e estão associados a menos efeitos colaterais.
As alilaminas, uma classe estruturalmente diferente de antifúngicos sintéticos, inibem uma etapa anterior na biossíntese do ergosterol. A alilamina mais comumente usada é a terbinafina (comercializada sob a marca Lamisil), que é usada topicamente para o tratamento de infecções dermatofíticas da pele, como pé de atleta, micose e coceira de atleta. O tratamento oral com terbinafina também é usado para o tratamento de fungos nas unhas das mãos e dos pés, mas pode estar associado ao raro efeito colateral da hepatotoxicidade.
Os polienos são uma classe de agentes antifúngicos produzidos naturalmente por certas bactérias actinomicetas do solo e estão estruturalmente relacionados aos macrolídeos. Essas moléculas grandes e lipofílicas se ligam ao ergosterol nas membranas citoplasmáticas dos fungos, criando poros. Exemplos comuns incluem nistatina e anfotericina B. A nistatina é normalmente usada como tratamento tópico para infecções fúngicas da pele, boca e vagina, mas também pode ser usada para infecções fúngicas intestinais. O medicamento anfotericina B é usado para infecções fúngicas sistêmicas, como aspergilose, meningite criptocócica, histoplasmose, blastomicose e candidíase. A anfotericina B foi a única droga antifúngica disponível por várias décadas, mas seu uso está associado a alguns efeitos colaterais graves, incluindo nefrotoxicidade (toxicidade renal).
A anfotericina B é frequentemente usada em combinação com a flucitosina, um análogo da pirimidina fluorada que é convertido por uma enzima específica para fungos em um produto tóxico que interfere na replicação do DNA e na síntese de proteínas em fungos. A flucitosina também está associada à hepatotoxicidade (toxicidade hepática) e à depressão da medula óssea.
Além de direcionar o ergosterol nas membranas celulares fúngicas, existem alguns medicamentos antifúngicos que têm como alvo outras estruturas fúngicas (Figura\(\PageIndex{2}\)). As equinocandinas, incluindo a caspofungina, são um grupo de compostos antifúngicos produzidos naturalmente que bloqueiam a síntese de β (1→3) glucano encontrado nas paredes celulares dos fungos, mas não encontrado nas células humanas. Essa classe de medicamentos tem o apelido de “penicilina para fungos”. A caspofungina é usada para o tratamento da aspergilose, bem como infecções sistêmicas por leveduras.
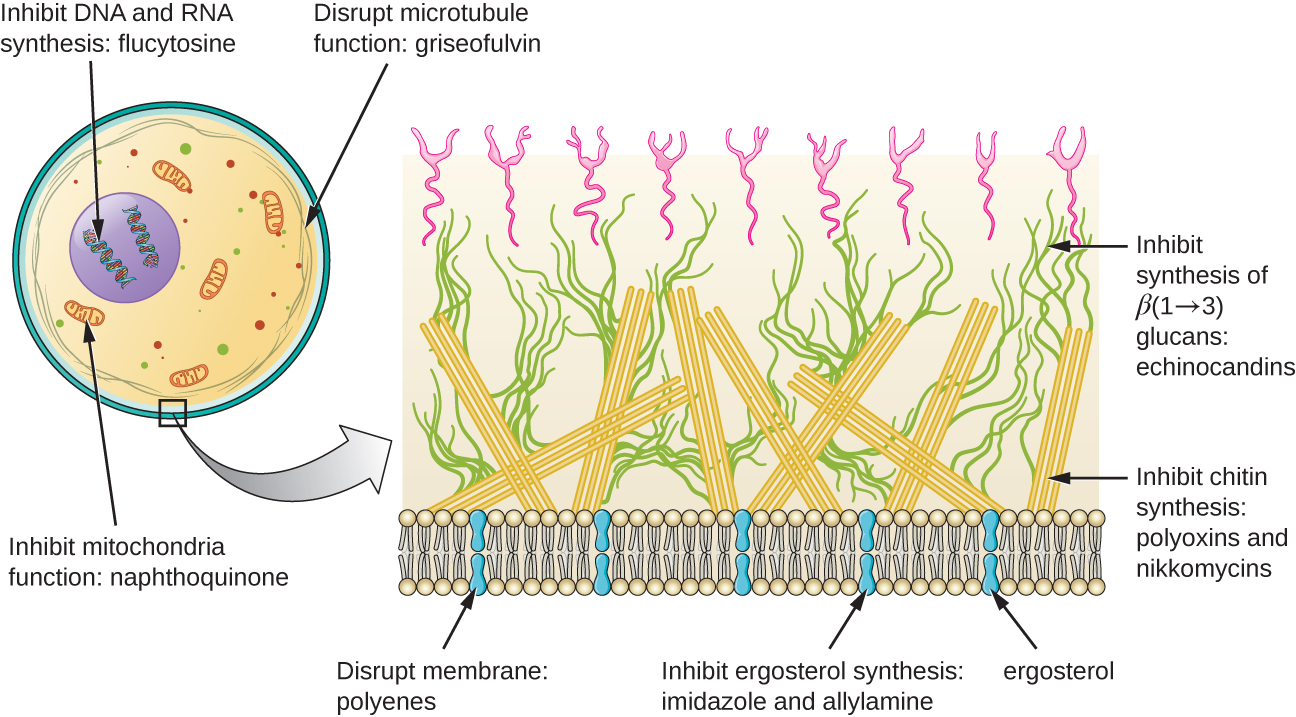
Embora a quitina seja apenas um constituinte menor das paredes celulares dos fungos, ela também está ausente nas células humanas, tornando-a um alvo seletivo. As polioxinas e as nikkomicinas são antifúngicos produzidos naturalmente que visam a síntese de quitina. As polioxinas são usadas para controlar fungos para fins agrícolas, e a nikkomicina Z está atualmente em desenvolvimento para uso em humanos no tratamento de infecções fúngicas e da febre do vale (coccidioidomicose), uma doença fúngica prevalente no sudoeste dos EUA. 1
Acredita-se que a griseofulvina antifúngica produzida naturalmente interrompa especificamente a divisão celular fúngica ao interferir nos microtúbulos envolvidos na formação do fuso durante a mitose. Foi um dos primeiros antifúngicos, mas seu uso está associado à hepatotoxicidade. Geralmente é administrado por via oral para tratar vários tipos de infecções dermatofíticas da pele quando outros tratamentos antifúngicos tópicos são ineficazes.
Existem alguns medicamentos que atuam como antimetabólitos contra processos fúngicos. Por exemplo, a atovaquona, representante da classe das naftoquinonas, é um antimetabólito semissintético para versões fúngicas e protozoárias de um citocromo mitocondrial importante no transporte de elétrons. Estruturalmente, é um análogo da coenzima Q, com a qual compete pela ligação eletrônica. É particularmente útil para o tratamento da pneumonia por Pneumocystis causada por Pneumocystis jirovecii. A combinação antibacteriana sulfametoxazol-trimetoprim também atua como antimetabólito contra P. jirovecii.
A tabela\(\PageIndex{1}\) mostra as várias classes terapêuticas de antifúngicos, categorizadas por modo de ação, com exemplos de cada uma.
| Mecanismo de ação | Classe de drogas | Medicamentos específicos | Usos clínicos |
|---|---|---|---|
| Inibir a síntese de ergosterol | Imidazóis | Miconazol, cetoconazol, clotrimazol | Infecções fúngicas da pele e infecções vaginais por fungos |
| Triazóis | Fluconazol | Infecções sistêmicas por fungos, candidíase oral e meningite criptocócica | |
| Alilaminas | Terbinafina | Infecções dermatofíticas da pele (pé de atleta, verme anelar, coceira de atleta) e infecções das unhas das mãos e dos pés | |
| Ligue o ergosterol na membrana celular e crie poros que rompem a membrana | Polienos | Nistatina | Usado topicamente para infecções fúngicas da pele, boca e vagina; também usado para infecções fúngicas do intestino |
| Anfotericina B | Várias infecções fúngicas sistêmicas | ||
| Inibir a síntese da parede celular | Equinocandinas | Caspofungina | Aspergilose e infecções sistêmicas por leveduras |
| Não aplicável | Nikcomicina Z | Coccidioidomicose (febre do vale) e infecções fúngicas | |
| Inibir os microtúbulos e a divisão celular | Não aplicável | Griseofulvina | Infecções dermatofíticas da pele |
Exercício\(\PageIndex{1}\)
Como a interrupção da biossíntese do ergosterol é um modo de ação eficaz para antifúngicos?
Tratamento de uma infecção fúngica dos pulmões
Jack, engenheiro de 48 anos, é HIV positivo, mas geralmente é saudável graças à terapia antirretroviral (ART). No entanto, após uma semana particularmente intensa de trabalho, ele desenvolveu febre e tosse seca. Ele presumiu que estava apenas resfriado ou gripe leve devido ao esforço excessivo e não pensou muito nisso. No entanto, após cerca de uma semana, ele começou a sentir fadiga, perda de peso e falta de ar. Ele decidiu visitar seu médico, que descobriu que Jack tinha um baixo nível de oxigenação no sangue. O médico solicitou exames de sangue, radiografia de tórax e coleta de uma amostra de escarro induzido para análise. Sua radiografia mostrou uma nebulosidade fina e várias pneumatoceles (bolsas de ar de paredes finas), o que indicava pneumonia por Pneumocystis (PCP), um tipo de pneumonia causada pelo fungo Pneumocystis jirovecii. O médico de Jack o internou no hospital e prescreveu Bactrim, uma combinação de sulfametoxazol e trimetoprim, para ser administrado por via intravenosa.
P. jirovecii é um fungo semelhante a uma levedura com um ciclo de vida semelhante ao dos protozoários. Dessa forma, foi classificado como protozoário até a década de 1980. Ele vive apenas no tecido pulmonar de pessoas infectadas e é transmitido de pessoa para pessoa, com muitas pessoas expostas quando crianças. Normalmente, P. jirovecii só causa pneumonia em indivíduos imunocomprometidos. Pessoas saudáveis podem carregar o fungo nos pulmões sem sintomas da doença. O PCP é particularmente problemático entre pacientes com HIV com sistema imunológico comprometido.
O PCP geralmente é tratado com Bactrim oral ou intravenoso, mas a atovaquona ou a pentamidina (outro medicamento antiparasitário) são alternativas. Se não for tratada, a PCP pode progredir, levando a um colapso pulmonar e quase 100% de mortalidade. Mesmo com a terapia antimicrobiana, o PCP ainda é responsável por 10% das mortes relacionadas ao HIV.
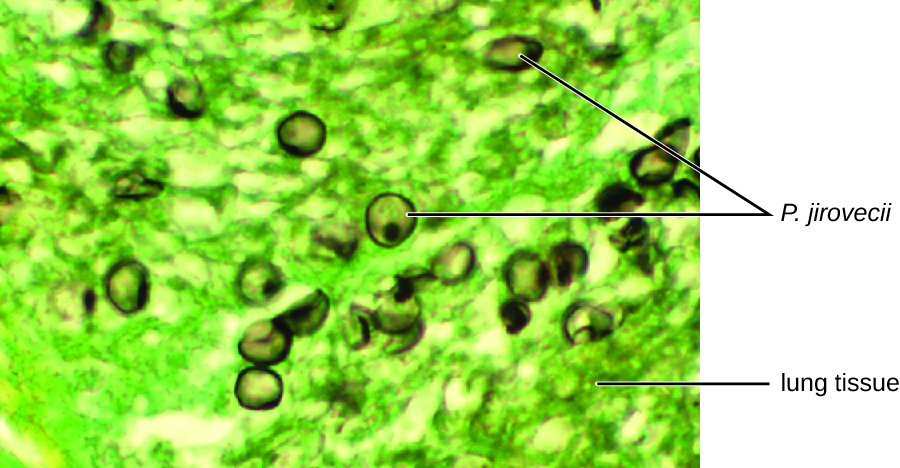
O exame citológico, usando ensaio de imunofluorescência direta (DFA), de um esfregaço da amostra de escarro de Jack confirmou a presença de P. jirovecii (Figura\(\PageIndex{3}\)). Além disso, os resultados dos exames de sangue de Jack revelaram que sua contagem de glóbulos brancos havia diminuído, tornando-o mais suscetível ao fungo. Seu médico revisou seu regime de TARV e fez ajustes. Depois de alguns dias de hospitalização, Jack foi liberado para continuar sua terapia antimicrobiana em casa. Com os ajustes em sua terapia ART, a contagem de CD4 de Jack começou a aumentar e ele conseguiu voltar ao trabalho.
Medicamentos antiprotozoários
Existem alguns mecanismos pelos quais os medicamentos antiprotozoários têm como alvo os protozoários infecciosos (Tabela\(\PageIndex{3}\)). Alguns são antimetabólitos, como atovaquona, proguanil e artemisininas. A atovaquona, além de ser antifúngica, bloqueia o transporte de elétrons em protozoários e é usada no tratamento de infecções por protozoários, incluindo malária, babesiose e toxoplasmose. O proguanil é outro antimetabólito sintético que é processado nas células parasitárias em sua forma ativa, o que inibe a síntese de ácido fólico por protozoários. É frequentemente usado em combinação com atovaquona, e a combinação é comercializada como malarona para tratamento e prevenção da malária.
A artemisinina, um antifúngico derivado de plantas descoberto pela primeira vez por cientistas chineses na década de 1970, é bastante eficaz contra a malária. Os derivados semissintéticos da artemisinina são mais solúveis em água do que a versão natural, o que os torna mais biodisponíveis. Embora o mecanismo de ação exato não seja claro, as artemisininas parecem atuar como pró-fármacos que são metabolizados pelas células-alvo para produzir espécies reativas de oxigênio (ROS) que danificam as células-alvo. Devido ao aumento da resistência aos medicamentos antimaláricos, as artemisininas também são comumente usadas em combinação com outros compostos antimaláricos na terapia combinada à base de artemisinina (ACT).
Vários antimetabólitos são usados para o tratamento da toxoplasmose causada pelo parasita Toxoplasma gondii. A sulfa sintética sulfadiazina inibe competitivamente uma enzima na produção de ácido fólico em parasitas e pode ser usada para tratar malária e toxoplasmose. A pirimetamina é uma droga sintética que inibe uma enzima diferente na via de produção do ácido fólico e é frequentemente usada em combinação com a sulfadoxina (outra droga sulfa) para o tratamento da malária ou em combinação com a sulfadiazina para o tratamento da toxoplasmose. Os efeitos colaterais da pirimetamina incluem diminuição da atividade da medula óssea que pode causar aumento de hematomas e baixa contagem de glóbulos vermelhos. Quando a toxicidade é preocupante, a espiramicina, um inibidor da síntese de proteínas macrolídeas, é normalmente administrada para o tratamento da toxoplasmose.
Duas classes de medicamentos antiprotozoários interferem na síntese de ácidos nucléicos: nitroimidazóis e quinolinas. Os nitroimidazóis, incluindo o metronidazol semissintético, que foi discutido anteriormente como um medicamento antibacteriano, e o tinidazol sintético, são úteis no combate a uma ampla variedade de patógenos protozoários, como Giardia lamblia, Entamoeba histolytica e Trichomonas vaginalis . Após a introdução nessas células em ambientes com baixo teor de oxigênio, os nitroimidazóis são ativados e introduzem a quebra da fita de DNA, interferindo na replicação do DNA nas células-alvo. Infelizmente, o metronidazol está associado à carcinogênese (desenvolvimento do câncer) em humanos.
Outro tipo de medicamento antiprotozoário sintético que há muito se acredita interferir especificamente na replicação do DNA em certos patógenos é a pentamidina. Historicamente, tem sido usado para o tratamento da doença do sono africana (causada pelo protozoário Trypanosoma brucei) e da leishmaniose (causada por protozoários do gênero Leishmania), mas também é um tratamento alternativo para o fungo Pneumocystis. Alguns estudos indicam que ele se liga especificamente ao DNA encontrado nos cinetoplastos (kDNA; estruturas longas semelhantes a mitocôndrias exclusivas dos tripanossomos), levando à clivagem do kDNA. No entanto, o DNA nuclear do parasita e do hospedeiro permanece inalterado. Também parece se ligar ao tRNA, inibindo a adição de aminoácidos ao tRNA, impedindo assim a síntese de proteínas. Os possíveis efeitos colaterais do uso de pentamidina incluem disfunção pancreática e danos ao fígado.
As quinolinas são uma classe de compostos sintéticos relacionados à quinina, que tem uma longa história de uso contra a malária. Acredita-se que as quinolinas interfiram na desintoxicação do heme, que é necessária para a decomposição efetiva da hemoglobina pelo parasita em aminoácidos dentro dos glóbulos vermelhos. Os derivados sintéticos cloroquina, quinacrina (também chamada de mepacrina) e mefloquina são comumente usados como antimaláricos, e a cloroquina também é usada para tratar a amebíase tipicamente causada pela Entamoeba histolytica. O uso profilático a longo prazo de cloroquina ou mefloquina pode resultar em efeitos colaterais graves, incluindo alucinações ou problemas cardíacos. Pacientes com deficiência de glicose-6-fosfato desidrogenase apresentam anemia grave quando tratados com cloroquina.
| Mecanismo de ação | Classe de drogas | Medicamentos específicos | Usos clínicos |
|---|---|---|---|
| Inibir o transporte de elétrons nas mitocôndrias | Naftoquinona | Atovaquona | Malária, babesiose e toxoplasmose |
| Inibir a síntese de ácido fólico | Não aplicável | Proquanil | Terapia combinada com atovaquona para tratamento e prevenção da malária |
| Sulfonamida | Sulfadiazina | Malária e toxoplasmose | |
| Não aplicável | Pirimetamina | Terapia combinada com sulfadoxina (fármaco sulfa) para malária | |
| Produz espécies reativas de oxigênio prejudiciais | Não aplicável | Artemisinina | Terapia combinada para tratar a malária |
| Iniba a síntese de DNA | Nitroimidazóis | Metronidazol, tinidazol | Infecções causadas por Giardia lamblia, Entamoeba histolytica e Trichomonas vaginalis |
| Não aplicável | Pentamidina | Doença do sono africana e leishmaniose | |
| Inibir a desintoxicação do heme | Quinolinas | Cloroquina | Malária e infecções por E. histolytica |
| Mepacrina, mefloquina | Malária |
Exercício\(\PageIndex{2}\)
Liste dois modos de ação dos medicamentos antiprotozoários.
Medicamentos antihelmínticos
Como os helmintos são eucariotos multicelulares como os humanos, desenvolver medicamentos com toxicidade seletiva contra eles é extremamente desafiador. Apesar disso, várias classes efetivas foram desenvolvidas (Tabela\(\PageIndex{3}\)). Os benzimidazóis sintéticos, como o mebendazol e o albendazol, ligam-se à β-tubulina helmíntica, impedindo a formação de microtúbulos. Os microtúbulos nas células intestinais dos vermes parecem ser particularmente afetados, levando a uma redução na absorção de glicose. Além de sua atividade contra uma ampla gama de helmintos, os benzimidazóis também são ativos contra muitos protozoários, fungos e vírus, e seu uso para inibir a mitose e a progressão do ciclo celular em células cancerosas está sendo estudado. 2 Os possíveis efeitos colaterais de seu uso incluem danos ao fígado e supressão da medula óssea.
As avermectinas são membros da família dos macrolídeos que foram descobertos pela primeira vez em um isolado de solo japonês, Streptomyces avermectinius. Um derivado semissintético mais potente da avermectina é a ivermectina, que se liga aos canais de cloreto dependentes do glutamato específicos dos invertebrados, incluindo helmintos, bloqueando a transmissão neuronal e causando fome, paralisia e morte dos vermes. A ivermectina é usada para tratar doenças de lombrigas, incluindo oncocercose (também chamada de cegueira dos rios, causada pelo verme Onchocerca volvulus) e estrongiloidíase (causada pelo verme Strongyloides stercoralis ou S. fuelleborni). A ivermectina também pode tratar insetos parasitas como ácaros, piolhos e percevejos, e não é tóxica para os humanos.
A niclosamida é uma droga sintética usada há mais de 50 anos para tratar infecções por tênias. Embora seu modo de ação não seja totalmente claro, a niclosamida parece inibir a formação de ATP em condições anaeróbicas e inibir a fosforilação oxidativa nas mitocôndrias de seus patógenos alvo. A niclosamida não é absorvida pelo trato gastrointestinal, portanto, pode atingir altas concentrações intestinais localizadas nos pacientes. Recentemente, foi demonstrado que também tem atividades antibacterianas, antivirais e antitumorais. 3 4 5
Outro medicamento anti-helmíntico sintético é o praziquantel, usado no tratamento de vermes parasitas e vermes do fígado, e é particularmente útil no tratamento da esquistossomose (causada por vermes sanguíneos de três gêneros de Schistosoma). Seu modo de ação ainda não está claro, mas parece causar o influxo de cálcio no verme, resultando em intenso espasmo e paralisia do verme. É frequentemente usado como uma alternativa preferida à niclosamida no tratamento de vermes quando o desconforto gastrointestinal limita o uso de niclosamida.
As tioxantenonas, outra classe de drogas sintéticas estruturalmente relacionadas à quinina, exibem atividade antiesquistossomal ao inibir a síntese de RNA. A tioxantenona lucantona e seu metabólito hycanthone foram os primeiros usados clinicamente, mas graves efeitos colaterais neurológicos, gastrointestinais, cardiovasculares e hepáticos levaram à sua descontinuação. A oxamniquina, um derivado menos tóxico do hycanthone, só é eficaz contra S. mansoni, uma das três espécies conhecidas por causar esquistossomose em humanos. O Praziquantel foi desenvolvido para atingir as outras duas espécies de esquistossomos, mas as preocupações com o aumento da resistência renovaram o interesse em desenvolver derivados adicionais da oxamniquina para atingir todas as três espécies de esquistossomos clinicamente importantes.
| Mecanismo de ação | Classe de drogas | Medicamentos específicos | Usos clínicos |
|---|---|---|---|
| Inibe a formação de microtúbulos, reduzindo a captação de glicose | Benzimidazóis | Mebendazol, albendazol | Variedade de infecções por helmintos |
| Bloqueie a transmissão neuronal, causando paralisia e inanição | Avermectinas | Ivermectina | Doenças da lombriga, incluindo cegueira dos rios e estrongiloidíase, e tratamento de insetos parasitas |
| Inibir a produção de ATP | Não aplicável | Niclosamida | Infecções por tênia intestinal |
| Induzir o influxo de cálcio | Não aplicável | Praziquantel | Esquistossomose (vermes do sangue) |
| Inibir a síntese de RNA | Tioxantenonas | Lucantona, hycanthone, oxamniquina | Esquistossomose (vermes do sangue) |
Exercício\(\PageIndex{3}\)
Por que os medicamentos anti-helmínticos são difíceis de desenvolver?
Medicamentos antivirais
Ao contrário da estrutura complexa de fungos, protozoários e helmintos, a estrutura viral é simples, consistindo em ácido nucléico, um revestimento proteico, enzimas virais e, às vezes, um envelope lipídico. Além disso, os vírus são patógenos intracelulares obrigatórios que usam a maquinaria celular do hospedeiro para se replicar. Essas características dificultam o desenvolvimento de medicamentos com toxicidade seletiva contra vírus.
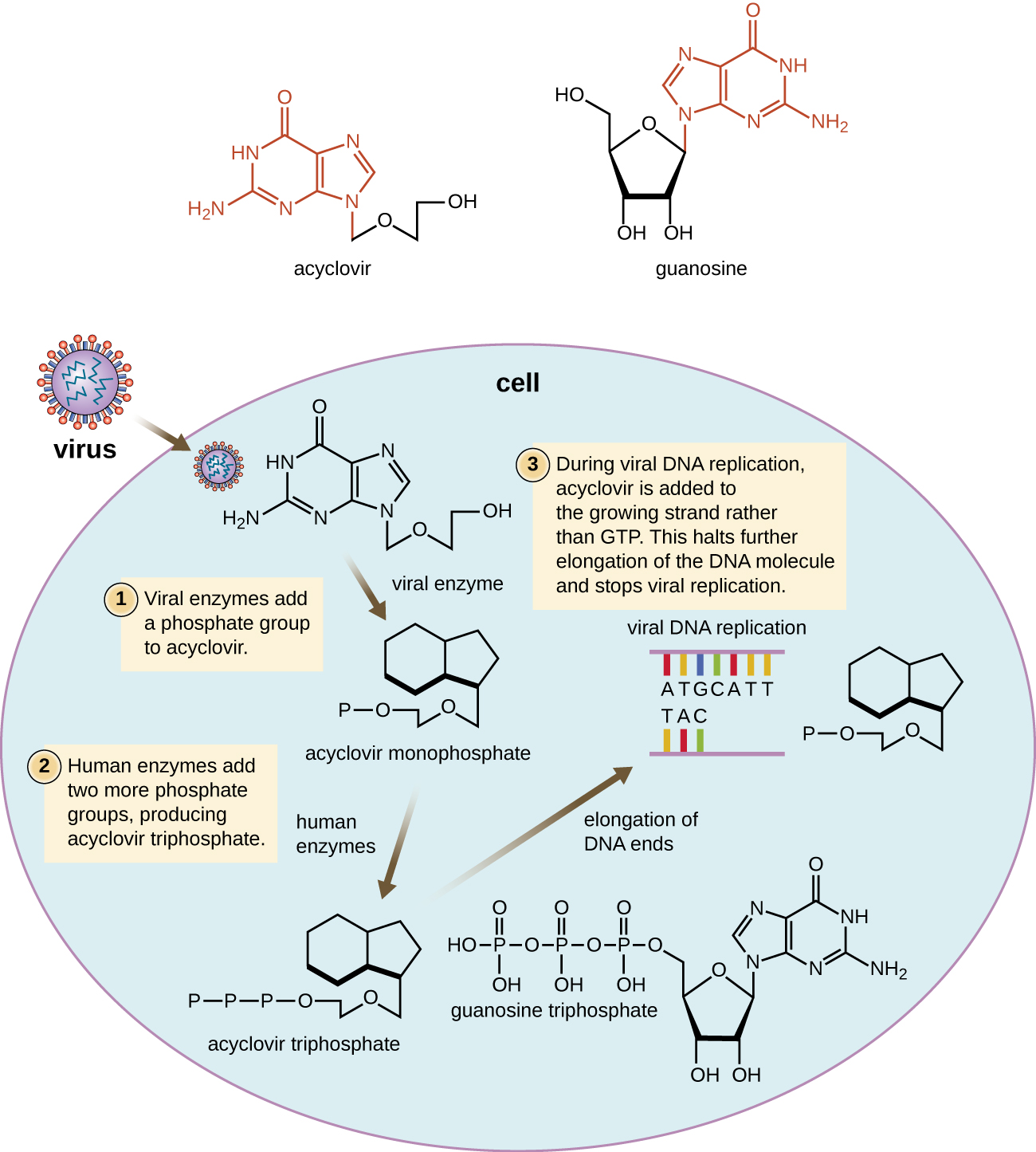
Muitos medicamentos antivirais são análogos de nucleosídeos e funcionam inibindo a biossíntese do ácido nucléico. Por exemplo, o aciclovir (comercializado como Zovirax) é um análogo sintético do nucleosídeo guanosina (Figura\(\PageIndex{4}\)). É ativado pela enzima viral timidina quinase do herpes simplex e, quando adicionado a uma fita de DNA em crescimento durante a replicação, causa o término da cadeia. Sua especificidade para células infectadas por vírus vem da necessidade de uma enzima viral para ativá-la e da maior afinidade da forma ativada pela DNA polimerase viral em comparação com a DNA polimerase da célula hospedeira. O aciclovir e seus derivados são frequentemente usados para o tratamento de infecções pelo vírus do herpes, incluindo herpes genital, varicela, herpes zoster, infecções pelo vírus Epstein-Barr e infecções por citomegalovírus. O aciclovir pode ser administrado de forma tópica ou sistêmica, dependendo da infecção. Um possível efeito colateral de seu uso inclui a nefrotoxicidade. O medicamento adenina-arabinosídeo, comercializado como vidarabina, é um análogo sintético da desoxiadenosina que tem um mecanismo de ação semelhante ao do aciclovir. Também é eficaz para o tratamento de vários vírus do herpes humano. No entanto, devido aos possíveis efeitos colaterais que envolvem baixa contagem de glóbulos brancos e neurotoxicidade, o tratamento com aciclovir agora é preferido.
A ribavirina, outro análogo sintético da guanosina, funciona por um mecanismo de ação que não está totalmente claro. Parece interferir na síntese de DNA e RNA, talvez reduzindo os pools intracelulares de trifosfato de guanosina (GTP). A ribavarina também parece inibir a RNA polimerase do vírus da hepatite C. É usado principalmente para o tratamento de vírus de RNA como hepatite C (em terapia combinada com interferon) e vírus sincicial respiratório. Os possíveis efeitos colaterais do uso de ribavirina incluem anemia e efeitos no desenvolvimento de crianças em gestação em pacientes grávidas. Nos últimos anos, outro análogo de nucleotídeo, o sofosbuvir (Solvaldi), também foi desenvolvido para o tratamento da hepatite C. O sofosbuvir é um análogo da uridina que interfere na atividade da polimerase viral. É comumente coadministrado com ribavirina, com e sem interferão.
A inibição da síntese de ácidos nucléicos não é o único alvo dos antivirais sintéticos. Embora o modo de ação da amantadina e sua relativa rimantadina não estejam totalmente esclarecidos, esses medicamentos parecem se ligar a uma proteína transmembranar que está envolvida na fuga do vírus da gripe dos endossomos. O bloqueio da fuga do vírus também impede a liberação de RNA viral nas células hospedeiras e a subsequente replicação viral. O aumento da resistência limitou o uso de amantadina e rimantadina no tratamento da gripe A. O uso de amantadina pode resultar em efeitos colaterais neurológicos, mas os efeitos colaterais da rimantadina parecem menos graves. Curiosamente, devido aos seus efeitos sobre substâncias químicas cerebrais, como dopamina e NMDA (N-metil D-aspartato), a amantadina e a rimantadina também são usadas para o tratamento da doença de Parkinson.
Os inibidores da neuraminidase, incluindo olsetamivir (Tamiflu), zanamivir (Relenza) e peramivir (Rapivab), visam especificamente os vírus da gripe, bloqueando a atividade da neuraminidase do vírus influenza, impedindo a liberação do vírus das células infectadas. Esses três antivirais podem diminuir os sintomas da gripe e encurtar a duração da doença, mas diferem em seus modos de administração: o olsetamivir é administrado por via oral, o zanamivir é inalado e o peramivir é administrado por via intravenosa. A resistência a esses inibidores da neuraminidase ainda parece ser mínima.
O pleconaril é um antiviral sintético em desenvolvimento que se mostrou promissor para o tratamento dos picornavírus. O uso de pleconaril para o tratamento do resfriado comum causado por rinovírus não foi aprovado pelo FDA em 2002 devido à falta de eficácia comprovada, falta de estabilidade e associação com menstruação irregular. Seu desenvolvimento posterior para esse fim foi interrompido em 2007. No entanto, o pleconaril ainda está sendo investigado para uso no tratamento de complicações fatais de enterovírus, como meningite e sepse. Também está sendo investigado para uso na erradicação global de um enterovírus específico, a poliomielite. 6 O pleconaril parece atuar ligando-se ao capsídeo viral e impedindo o desrevestimento de partículas virais dentro das células hospedeiras durante a infecção viral.
Vírus com ciclos de vida complexos, como o HIV, podem ser mais difíceis de tratar. Primeiro, o HIV tem como alvo os glóbulos brancos CD4 positivos, que são necessários para uma resposta imune normal à infecção. Em segundo lugar, o HIV é um retrovírus, o que significa que ele converte seu genoma de RNA em uma cópia de DNA que se integra ao genoma da célula hospedeira, escondendo-se assim no DNA da célula hospedeira. Terceiro, a transcriptase reversa do HIV carece de atividade de revisão e introduz mutações que permitem o rápido desenvolvimento da resistência a medicamentos antivirais. Para ajudar a evitar o surgimento de resistência, uma combinação de medicamentos antivirais sintéticos específicos é normalmente usada na TARV para o HIV (Figura\(\PageIndex{5}\)).
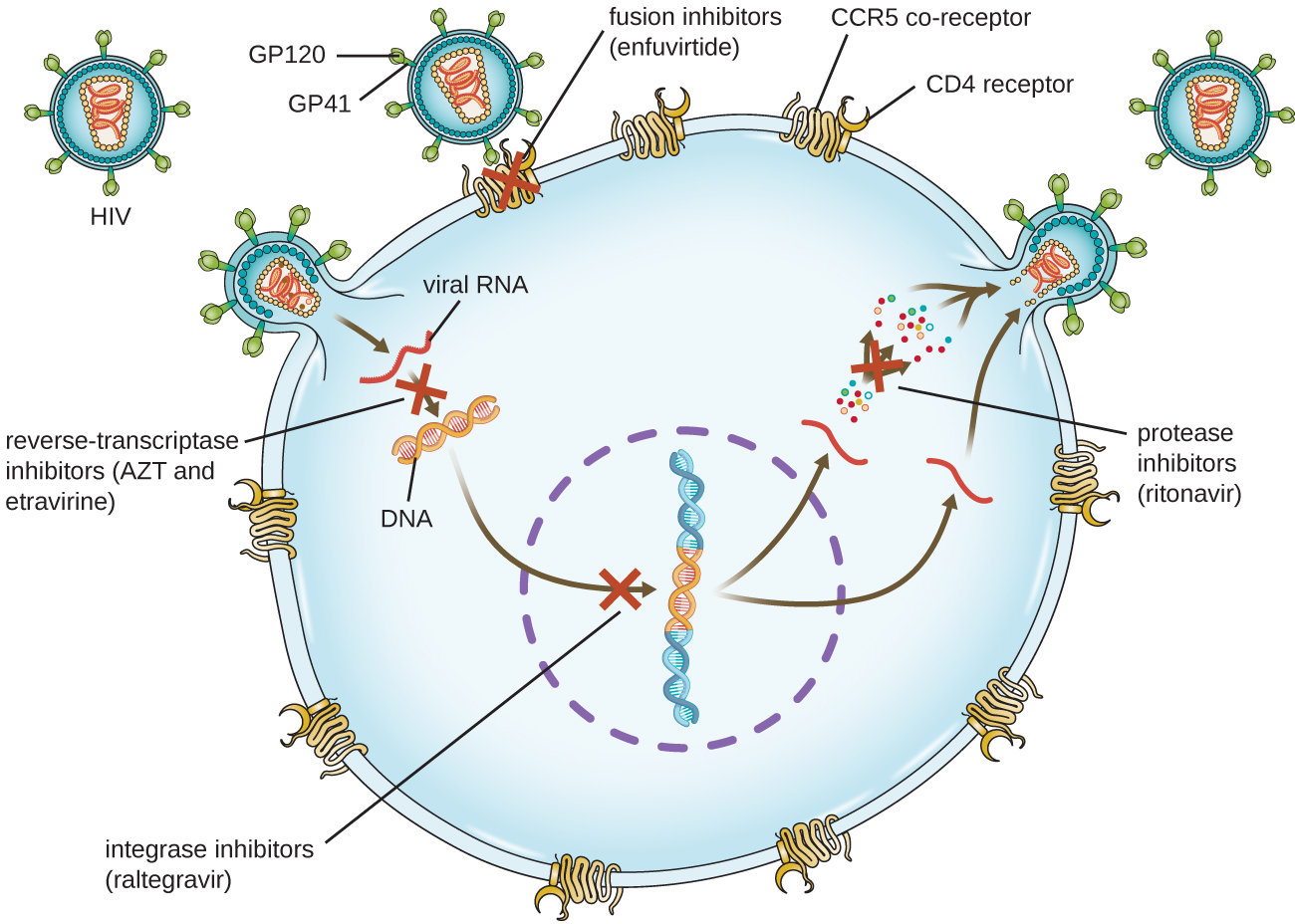
Os inibidores da transcriptase reversa bloqueiam a etapa inicial da conversão do genoma do RNA viral em DNA e podem incluir inibidores analógicos de nucleosídeos competitivos (por exemplo, azidotimidina/zidovudina ou AZT) e inibidores não competitivos não nucleosídeos (por exemplo, etravirina) que se ligam à transcriptase reversa e causam uma inativando a mudança conformacional. Medicamentos chamados inibidores de protease (por exemplo, ritonavir) bloqueiam o processamento de proteínas virais e previnem a maturação viral. Os inibidores da protease também estão sendo desenvolvidos para o tratamento de outros tipos virais. 7 Por exemplo, o simeprevir (Olysio) foi aprovado para o tratamento da hepatite C e é administrado com ribavirina e interferon em terapia combinada. Os inibidores da integrase (por exemplo, raltegravir) bloqueiam a atividade da integrase do HIV responsável pela recombinação de uma cópia de DNA do genoma viral no cromossomo da célula hospedeira. Classes adicionais de medicamentos para o tratamento do HIV incluem os antagonistas do CCR5 e os inibidores de fusão (por exemplo, enfuviritida), que impedem a ligação do HIV ao correceptor da célula hospedeira (receptor de quimiocina tipo 5 [CCR5]) e a fusão do envelope viral com a membrana da célula hospedeira, respectivamente. A tabela\(\PageIndex{4}\) mostra as várias classes terapêuticas de medicamentos antivirais, categorizadas por modo de ação, com exemplos de cada uma.
| Mecanismo de ação | Medicamento | Usos clínicos |
|---|---|---|
| Inibição análoga de nucleósido da síntese de ácido nucleico | Aciclovir | Infecções pelo vírus herpes |
| Azidotimidina/zidovudina (AZT) | infecções por HIV | |
| Ribavirina | Infecções pelo vírus da hepatite C e pelo vírus sincicial respiratório | |
| Vidarabina | Infecções pelo vírus herpes | |
| Sofosbuvir | infecções pelo vírus da hepatite C | |
| Inibição não competitiva não nucleósida | Etravirina | infecções por HIV |
| Inibir a fuga do vírus dos endossomos | Amantadina, rimantadina | Infecções pelo vírus da gripe |
| Inibir a neuraminadase | Olsetamivir, zanamivir, peramivir | Infecções pelo vírus da gripe |
| Inibir o desrevestimento viral | Pleconaril | Infecções graves por enterovírus |
| Inibição da protease | Ritonavir | infecções por HIV |
| Simeprevir | infecções pelo vírus da hepatite C | |
| Inibição da integrase | Raltegravir | infecções por HIV |
| Inibição da fusão de membrana | Enfuviritida | infecções por HIV |
Exercício\(\PageIndex{4}\)
Por que o HIV é difícil de tratar com antivirais?
Para saber mais sobre as várias classes de medicamentos antirretrovirais usados na TARV da infecção pelo HIV, explore cada um dos medicamentos nas classes de medicamentos para HIV fornecidas pelo Departamento de Saúde e Serviços Humanos dos EUA neste site.
Conceitos principais e resumo
- Como fungos, protozoários e helmintos são organismos eucarióticos como as células humanas, é mais difícil desenvolver medicamentos antimicrobianos que os visem especificamente. Da mesma forma, é difícil atacar vírus porque os vírus humanos se replicam dentro das células humanas.
- Os medicamentos antifúngicos interferem na síntese do ergosterol, se ligam ao ergosterol para interromper a integridade da membrana celular fúngica ou têm como alvo componentes específicos da parede celular ou outras proteínas celulares.
- Os medicamentos antiprotozoários aumentam os níveis celulares das espécies reativas de oxigênio, interferem na replicação do DNA protozoário (nuclear versus kDNA, respectivamente) e interrompem a desintoxicação do heme.
- Os medicamentos anti-helmínticos interrompem a formação de microtúbulos helmínticos e protozoários; bloqueiam as transmissões neuronais; inibem a formação anaeróbica de ATP e/ou a fosforilação oxidativa; induzem um influxo de cálcio nas tênias, causando espasmos e paralisia; e interferem na síntese de RNA nos esquistossomos.
- Os medicamentos antivirais inibem a entrada viral, inibem o desrevestimento viral, inibem a biossíntese do ácido nucléico, previnem a fuga viral dos endossomos nas células hospedeiras e previnem a liberação viral das células infectadas.
- Como pode facilmente sofrer mutações para se tornar resistente a medicamentos, o HIV é normalmente tratado com uma combinação de vários medicamentos antirretrovirais, que podem incluir inibidores da transcriptase reversa, inibidores da protease, inibidores da integrase e medicamentos que interferem na ligação viral e na fusão de iniciar a infecção.
Notas de pé
- 1 Centros de Controle e Prevenção de Doenças. “Febre do vale: a conscientização é fundamental.” www.cdc.gov/features/valleyfever/. Acessado em 1º de junho de 2016.
- 2 B. Chu et al. “Um derivado do benzimidazol com atividade antitumoral bloqueia a atividade do EGFR e do HER2 e regula positivamente o DR5 nas células de câncer de mama.” Morte celular e doença 6 (2015) :e1686
- 3 J.-X. Pan e cols.. “A niclosamida, um antigo agente anti-helmíntico, demonstra atividade antitumoral bloqueando várias vias de sinalização das células-tronco cancerosas.” Jornal Chinês do Câncer 31, nº 4 (2012) :178—184.
- 4 F. Imperi et al. “Nova vida para um medicamento antigo: o medicamento anti-helmíntico niclosamida inibe a detecção de quórum de Pseudomonas aeruginosa”. Agentes antimicrobianos e quimioterapia 57 no. 2 (2013) :996-1005.
- 5 A. Jurgeit et al. “A niclosamida é um transportador de prótons e tem como alvo endossomos ácidos com amplos efeitos antivirais.” Patógenos PLoS 8 no. 10 (2012) :e1002976.
- 6 M. J. Abzug. “Os enterovírus: problemas que precisam de tratamentos.” Jornal da Infecção 68 no. S1 (2014) :108—14.
- 7 B.L. Pearlman. “Inibidores de protease para o tratamento da infecção crônica pelo genótipo 1 da hepatite C: o novo padrão de tratamento”. Doenças Infecciosas de Lancet 12 no. 9 (2012) :717—728.