
12.2: Visualizando e caracterizando o DNA
- Page ID
- 181715

Objetivos de
- Explicar o uso de sondas de ácido nucléico para visualizar sequências específicas de DNA
- Explicar o uso da eletroforese em gel para separar fragmentos de DNA
- Explicar o princípio da restrição, análise do polimorfismo do comprimento do fragmento e seus usos
- Compare e contraste manchas do sul e do norte
- Explicar os princípios e usos da análise de microarray
- Descreva os métodos usados para separar e visualizar variantes de proteína
- Explicar o método e os usos da reação em cadeia da polimerase e do sequenciamento de DNA
A sequência de uma molécula de DNA pode nos ajudar a identificar um organismo quando comparada às sequências conhecidas alojadas em um banco de dados. A sequência também pode nos dizer algo sobre a função de uma parte específica do DNA, como se ela codifica uma proteína específica. Comparar assinaturas de proteínas - os níveis de expressão de matrizes específicas de proteínas - entre amostras é um método importante para avaliar as respostas celulares a uma infinidade de fatores ambientais e estresses. A análise das assinaturas de proteínas pode revelar a identidade de um organismo ou como uma célula está respondendo durante a doença.
O DNA e as proteínas de interesse são microscópicos e normalmente misturados com muitas outras moléculas, incluindo DNA ou proteínas, irrelevantes para nossos interesses. Muitas técnicas foram desenvolvidas para isolar e caracterizar moléculas de interesse. Esses métodos foram originalmente desenvolvidos para fins de pesquisa, mas em muitos casos foram simplificados a ponto de o uso clínico rotineiro ser possível. Por exemplo, muitos patógenos, como a bactéria Helicobacter pylori, que causa úlceras estomacais, podem ser detectados por meio de testes baseados em proteínas. Além disso, um número crescente de ensaios de identificação altamente específicos e precisos baseados em amplificação de DNA agora pode detectar patógenos como bactérias entéricas resistentes a antibióticos, vírus herpes simplex, vírus varicela-zoster e muitos outros.
Análise molecular do DNA
Nesta subseção, descreveremos alguns dos métodos básicos usados para separar e visualizar fragmentos específicos de DNA que são do interesse de um cientista. Alguns desses métodos não exigem conhecimento da sequência completa da molécula de DNA. Antes do advento do sequenciamento rápido de DNA, esses métodos eram os únicos disponíveis para trabalhar com o DNA, mas ainda formam o arsenal básico de ferramentas usadas pelos geneticistas moleculares para estudar as respostas do corpo às doenças microbianas e outras.
Sondagem de ácido nucléico
As moléculas de DNA são pequenas e as informações contidas em sua sequência são invisíveis. Como um pesquisador isola um trecho específico de DNA ou, ao isolá-lo, determina de que organismo ele é, qual é sua sequência ou qual é sua função? Um método para identificar a presença de uma determinada sequência de DNA usa pedaços de DNA construídos artificialmente, chamados de sondas. As sondas podem ser usadas para identificar diferentes espécies bacterianas no ambiente e muitas sondas de DNA estão agora disponíveis para detectar patógenos clinicamente. Por exemplo, sondas de DNA são usadas para detectar os patógenos vaginais Candida albicans, Gardnerella vaginalis e Trichomonas vaginalis.
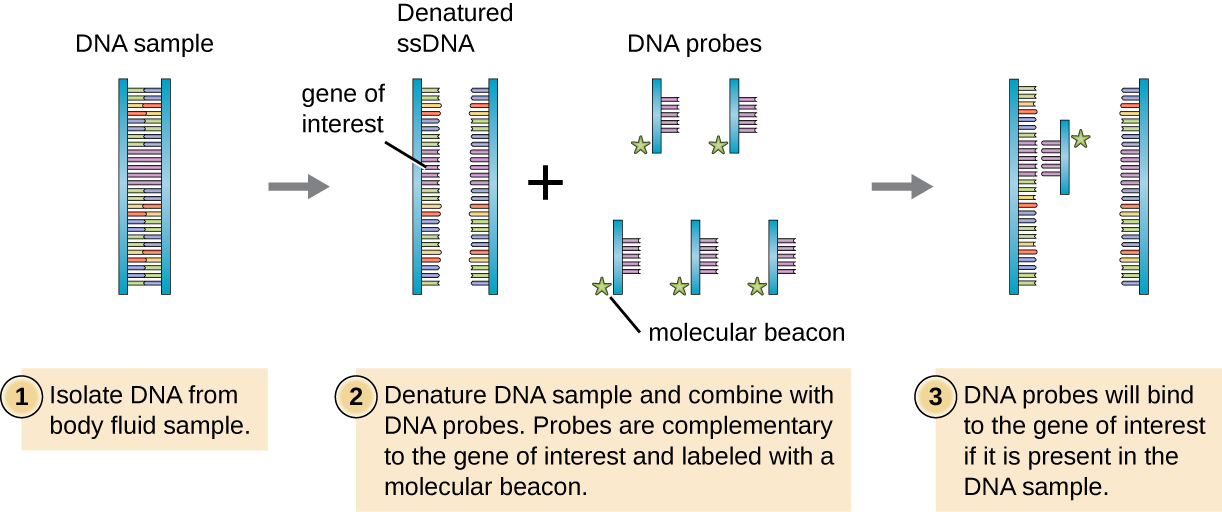
Para rastrear uma biblioteca genômica para um determinado gene ou sequência de interesse, os pesquisadores devem saber algo sobre esse gene. Se os pesquisadores tiverem uma parte da sequência de DNA para o gene de interesse, eles podem projetar uma sonda de DNA, um fragmento de DNA de fita simples que é complementar a parte do gene de interesse e diferente de outras sequências de DNA na amostra. A sonda de DNA pode ser sintetizada quimicamente por laboratórios comerciais ou pode ser criada clonando, isolando e desnaturando um fragmento de DNA de um organismo vivo. Em ambos os casos, a sonda de DNA deve ser rotulada com uma etiqueta molecular ou farol, como um átomo de fósforo radioativo (como é usado para autorradiografia) ou um corante fluorescente (como é usado na hibridização fluorescente in situ, ou FISH), para que a sonda e o DNA ao qual ela se liga possam ser vistos (Figura \(\PageIndex{1}\)). A amostra de DNA que está sendo sondada também deve ser desnaturada para torná-la de fita simples, para que a sonda de DNA de fita simples possa se recolocar na amostra de DNA de fita simples em locais onde suas sequências são complementares. Embora essas técnicas sejam valiosas para o diagnóstico, seu uso direto no escarro e em outras amostras corporais pode ser problemático devido à natureza complexa dessas amostras. O DNA geralmente deve primeiro ser isolado de amostras corporais por meio de métodos de extração química antes que uma sonda de DNA possa ser usada para identificar patógenos.
Foco clínico: Parte 2
Os sintomas leves, semelhantes aos da gripe, que Kayla está sentindo podem ser causados por vários agentes infecciosos. Além disso, várias doenças autoimunes não infecciosas, como esclerose múltipla, lúpus eritematoso sistêmico (LES) e esclerose lateral amiotrófica (ELA), também apresentam sintomas consistentes com os primeiros sintomas de Kayla. No entanto, ao longo de várias semanas, os sintomas de Kayla pioraram. Ela começou a sentir dores nas articulações nos joelhos, palpitações cardíacas e uma estranha mancha nos músculos faciais. Além disso, ela sofria de rigidez no pescoço e dores de cabeça dolorosas. Com relutância, ela decidiu que era hora de procurar atendimento médico.
Exercício\(\PageIndex{1}\)
- Os novos sintomas de Kayla fornecem alguma pista sobre o tipo de infecção ou outra condição médica que ela possa ter?
- Quais testes ou ferramentas um profissional de saúde pode usar para identificar o patógeno causador dos sintomas de Kayla?
Eletroforese em gel de agarose
Há várias situações em que um pesquisador pode querer separar fisicamente uma coleção de fragmentos de DNA de tamanhos diferentes. Um pesquisador também pode digerir uma amostra de DNA com uma enzima de restrição para formar fragmentos. O tamanho resultante e o padrão de distribuição de fragmentos geralmente podem fornecer informações úteis sobre a sequência de bases de DNA que podem ser usadas, assim como uma leitura de código de barras, para identificar o indivíduo ou a espécie à qual o DNA pertence.
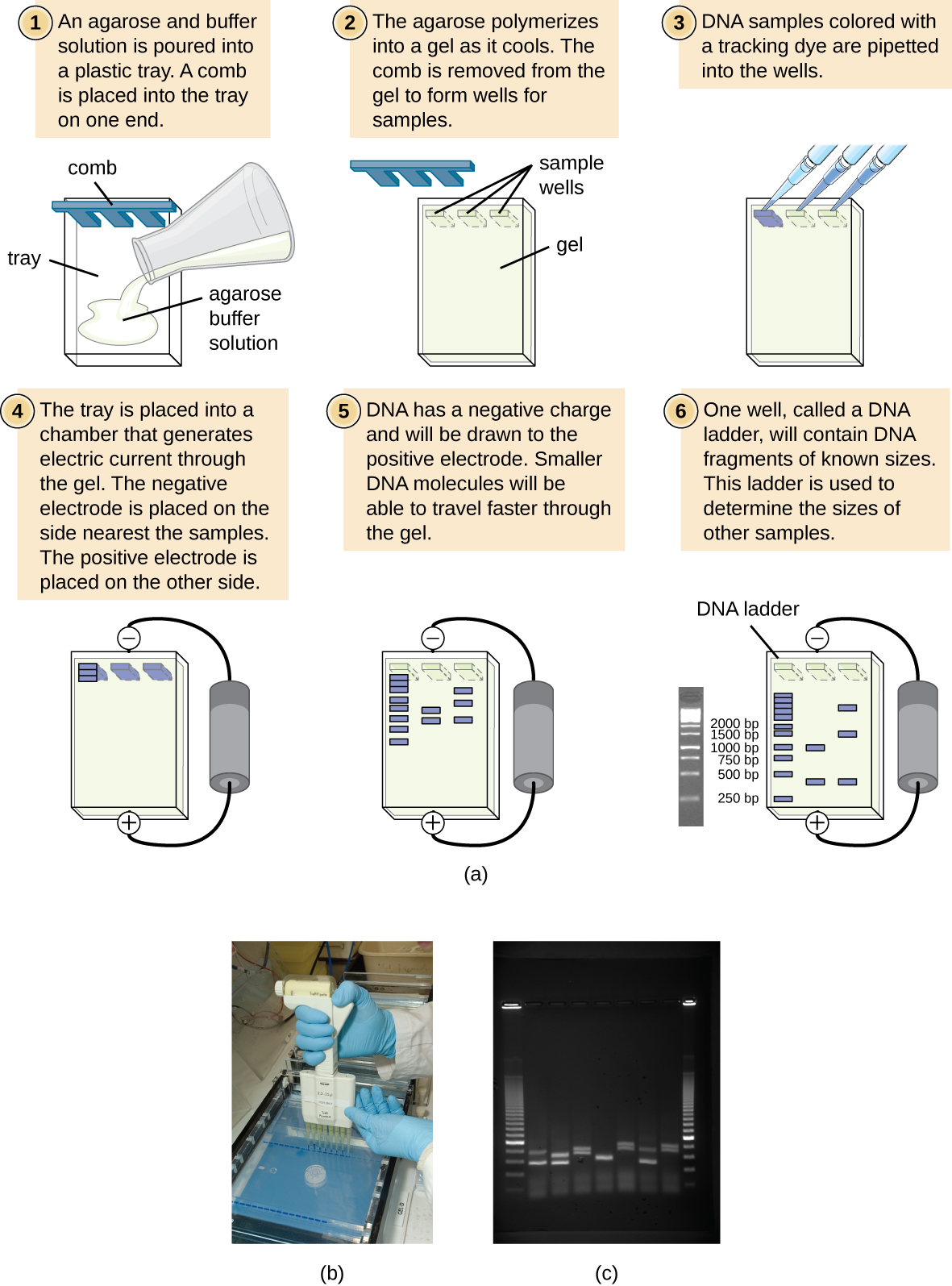
A eletroforese em gel é uma técnica comumente usada para separar moléculas biológicas com base no tamanho e nas características bioquímicas, como carga e polaridade. A eletroforese em gel de agarose é amplamente usada para separar DNA (ou RNA) de tamanhos variados que podem ser gerados pela digestão enzimática de restrição ou por outros meios, como a PCR (Figura\(\PageIndex{2}\)).
Devido à sua coluna vertebral carregada negativamente, o DNA é fortemente atraído por um eletrodo positivo. Na eletroforese em gel de agarose, o gel é orientado horizontalmente em uma solução tampão. As amostras são carregadas em poços de amostra na lateral do gel mais próxima do eletrodo negativo e, em seguida, puxadas pela peneira molecular da matriz de agarose em direção ao eletrodo positivo. A matriz de agarose impede o movimento de moléculas maiores através do gel, enquanto moléculas menores passam mais facilmente. Assim, a distância de migração está inversamente correlacionada ao tamanho do fragmento de DNA, com fragmentos menores percorrendo uma distância maior através do gel. Os tamanhos dos fragmentos de DNA em uma amostra podem ser estimados em comparação com fragmentos de tamanho conhecido em uma escada de DNA que também funcionam no mesmo gel. Para separar fragmentos de DNA muito grandes, como cromossomos ou genomas virais, a eletroforese em gel de agarose pode ser modificada alternando periodicamente a orientação do campo elétrico durante a eletroforese em gel de campo pulsado (PFGE). No PFGE, fragmentos menores podem se reorientar e migrar um pouco mais rápido do que fragmentos maiores e essa técnica pode, portanto, servir para separar fragmentos muito grandes que, de outra forma, viajariam juntos durante a eletroforese padrão em gel de agarose. Em qualquer uma dessas técnicas de eletroforese, a localização dos fragmentos de DNA ou RNA no gel pode ser detectada por vários métodos. Um método comum é adicionar brometo de etídio, uma mancha que se insere nos ácidos nucléicos em locais não específicos e pode ser visualizada quando exposta à luz ultravioleta. Outras manchas que são mais seguras do que o brometo de etídio, um potencial cancerígeno, estão agora disponíveis.
Análise de polimorfismo de comprimento de fragmento de restrição (RFLP)
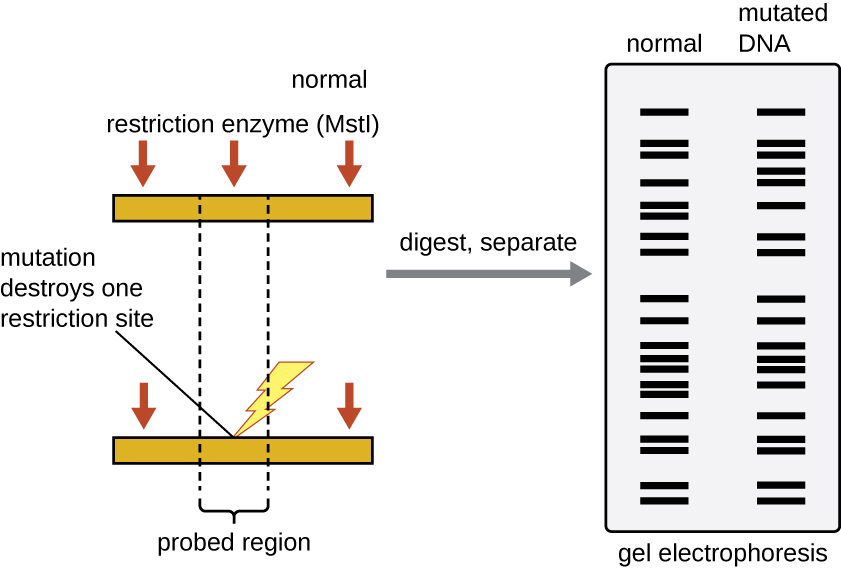
Os locais de reconhecimento de enzimas de restrição são curtos (apenas alguns nucleotídeos), palíndromos específicos da sequência e podem ser encontrados em todo o genoma. Assim, diferenças nas sequências de DNA nos genomas dos indivíduos levarão a diferenças na distribuição dos locais de reconhecimento de enzimas de restrição que podem ser visualizados como padrões distintos de bandas em um gel após a eletroforese em gel de agarose. A análise do polimorfismo de comprimento do fragmento de restrição (RFLP) compara os padrões de bandas de DNA de diferentes amostras de DNA após a digestão por restrição (Figura\(\PageIndex{3}\)).
A análise de RFLP tem muitas aplicações práticas na medicina e na ciência forense. Por exemplo, epidemiologistas usam a análise de RFLP para rastrear e identificar a origem de microrganismos específicos implicados em surtos de intoxicação alimentar ou certas doenças infecciosas. A análise de RFLP também pode ser usada no DNA humano para determinar padrões de herança de cromossomos com genes variantes, incluindo aqueles associados a doenças hereditárias ou para estabelecer a paternidade.
Cientistas forenses usam a análise RFLP como uma forma de impressão digital de DNA, que é útil para analisar o DNA obtido de cenas de crimes, suspeitos e vítimas. Amostras de DNA são coletadas, o número de cópias das moléculas de DNA da amostra é aumentado usando PCR e, em seguida, submetido à digestão enzimática de restrição e eletroforese em gel de agarose para gerar padrões de bandas específicos. Ao comparar os padrões de bandas das amostras coletadas na cena do crime com as coletadas de suspeitos ou vítimas, os investigadores podem determinar definitivamente se as evidências de DNA coletadas no local foram deixadas para trás por suspeitos ou vítimas.
Southern Blots e modificações
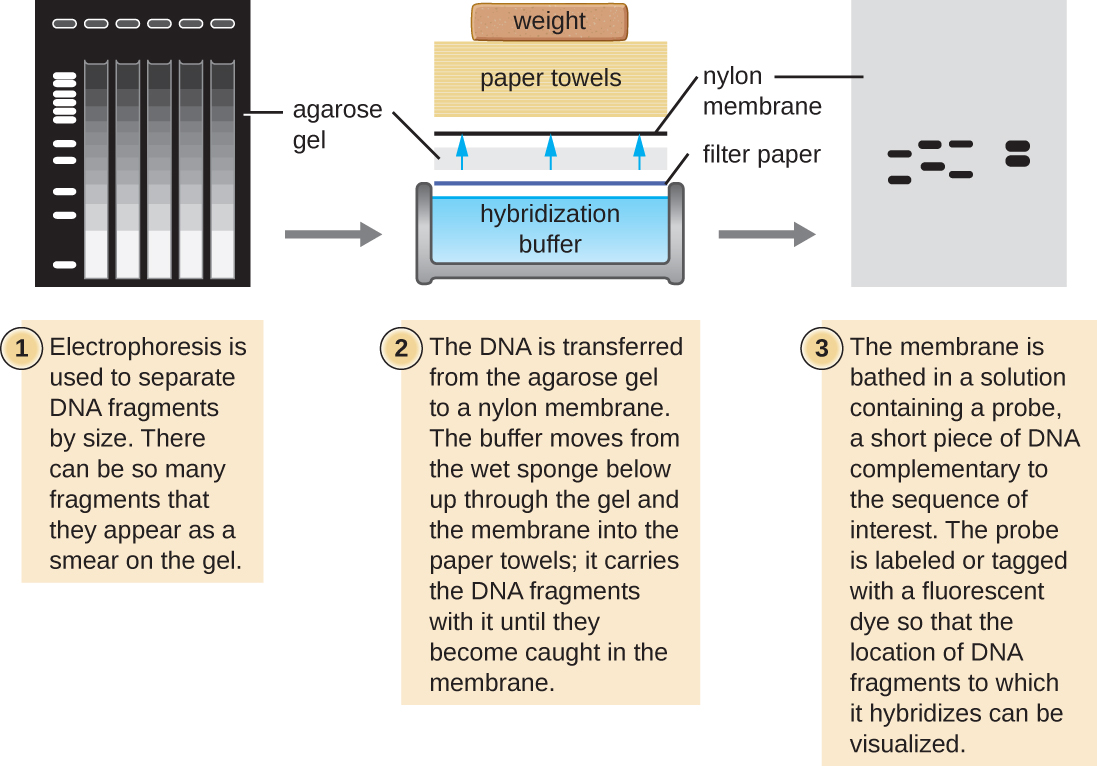
Várias técnicas moleculares aproveitam a complementaridade de sequências e a hibridização entre os ácidos nucléicos de uma amostra e as sondas de DNA. Normalmente, a sondagem de amostras de ácido nucléico dentro de um gel não é bem-sucedida porque, à medida que a sonda de DNA penetra em um gel, os ácidos nucléicos da amostra dentro do gel se difundem. Assim, técnicas de borrão são comumente usadas para transferir ácidos nucléicos para uma membrana fina e com carga positiva feita de nitrocelulose ou náilon. Na técnica Southern blot, desenvolvida por Sir Edwin Southern em 1975, os fragmentos de DNA dentro de uma amostra são primeiro separados por eletroforese em gel de agarose e depois transferidos para uma membrana por ação capilar (Figura\(\PageIndex{4}\)). Os fragmentos de DNA que se ligam à superfície da membrana são então expostos a uma sonda de DNA específica de fita simples marcada com um farol molecular radioativo ou fluorescente para auxiliar na detecção. Southern blots podem ser usados para detectar a presença de certas sequências de DNA em uma determinada amostra de DNA. Depois que o DNA alvo dentro da membrana é visualizado, os pesquisadores podem cortar a parte da membrana que contém o fragmento para recuperar o fragmento de DNA de interesse.
Variações da mancha do Sul - a mancha de pontos, a mancha de fenda e a mancha - não envolvem eletroforese, mas concentram o DNA de uma amostra em um pequeno local em uma membrana. Após a hibridização com uma sonda de DNA, a intensidade do sinal detectado é medida, permitindo ao pesquisador estimar a quantidade de DNA alvo presente na amostra.
Uma mancha de colônia é outra variação da mancha sul na qual colônias representando diferentes clones em uma biblioteca genômica são transferidas para uma membrana pressionando a membrana na placa de cultura. As células da membrana são lisadas e a membrana pode então ser sondada para determinar quais colônias dentro de uma biblioteca genômica abrigam o gene alvo. Como as colônias na placa ainda estão crescendo, as células de interesse podem ser isoladas da placa.
Na mancha norte, outra variação da mancha sul, o RNA (não o DNA) é imobilizado na membrana e sondado. As manchas do norte são normalmente usadas para detectar a quantidade de mRNA produzida por meio da expressão gênica em uma amostra de tecido ou organismo.
Análise de microarray
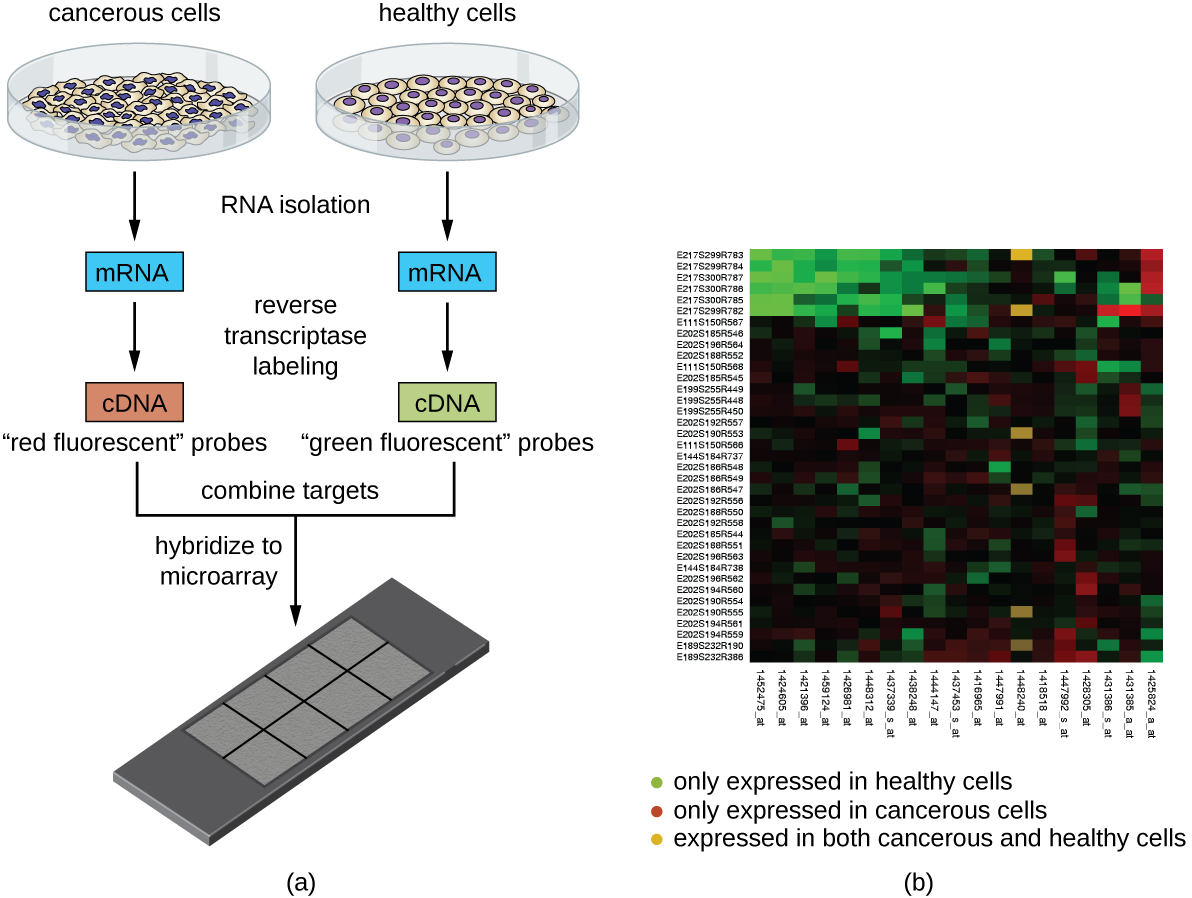
Outra técnica que capitaliza a hibridização entre sequências complementares de ácidos nucléicos é chamada de análise de microarray. A análise de microarray é útil para a comparação de padrões de expressão gênica entre diferentes tipos de células — por exemplo, células infectadas com um vírus versus células não infectadas ou células cancerosas versus células saudáveis (Figura\(\PageIndex{5}\)).
Normalmente, o DNA ou cDNA de uma amostra experimental é depositado em uma lâmina de vidro ao lado de sequências de DNA conhecidas. Cada lâmina pode conter mais de 30.000 tipos diferentes de fragmentos de DNA. Fragmentos de DNA distintos (abrangendo toda a biblioteca genômica de um organismo) ou fragmentos de cDNA (correspondentes ao conjunto completo de genes expressos de um organismo) podem ser identificados individualmente em uma lâmina de vidro.
Uma vez depositado na lâmina, o DNA genômico ou o mRNA podem ser isolados das duas amostras para comparação. Se o mRNA for isolado, ele é transcrito reversamente para cDNA usando transcriptase reversa. Em seguida, as duas amostras de DNA genômico ou cDNA são marcadas com diferentes corantes fluorescentes (normalmente vermelho e verde). As amostras de DNA genômico marcadas são então combinadas em quantidades iguais, adicionadas ao chip do microarray e permitidas a hibridização em pontos complementares no microarray.
A hibridização das moléculas de DNA genômico da amostra pode ser monitorada medindo a intensidade da fluorescência em pontos específicos do microarray. Diferenças na quantidade de hibridização entre as amostras podem ser facilmente observadas. Se apenas os ácidos nucléicos de uma amostra hibridizarem para um ponto específico no microarray, esse ponto aparecerá verde ou vermelho. No entanto, se os ácidos nucléicos de ambas as amostras hibridizarem, a mancha aparecerá amarela devido à combinação dos corantes vermelho e verde.
Embora a tecnologia de microarray permita uma comparação holística entre duas amostras em pouco tempo, ela requer equipamentos de detecção e software de análise sofisticados (e caros). Por causa das despesas, essa tecnologia normalmente se limita aos ambientes de pesquisa. Pesquisadores usaram a análise de microarray para estudar como a expressão gênica é afetada em organismos infectados por bactérias ou vírus ou submetidos a certos tratamentos químicos.
Explore a tecnologia de microchip neste site interativo.
Exercício\(\PageIndex{2}\)
- Em que consiste uma sonda de DNA?
- Por que um Southern blot é usado após a eletroforese em gel de um digestão de DNA?
Análise Molecular de Proteínas
Em muitos casos, pode não ser desejável ou possível estudar DNA ou RNA diretamente. As proteínas podem fornecer informações específicas da espécie para identificação, bem como informações importantes sobre como e se uma célula ou tecido está respondendo à presença de um microrganismo patogênico. Várias proteínas requerem métodos diferentes de isolamento e caracterização.
Eletroforese em gel de poliacrilamida
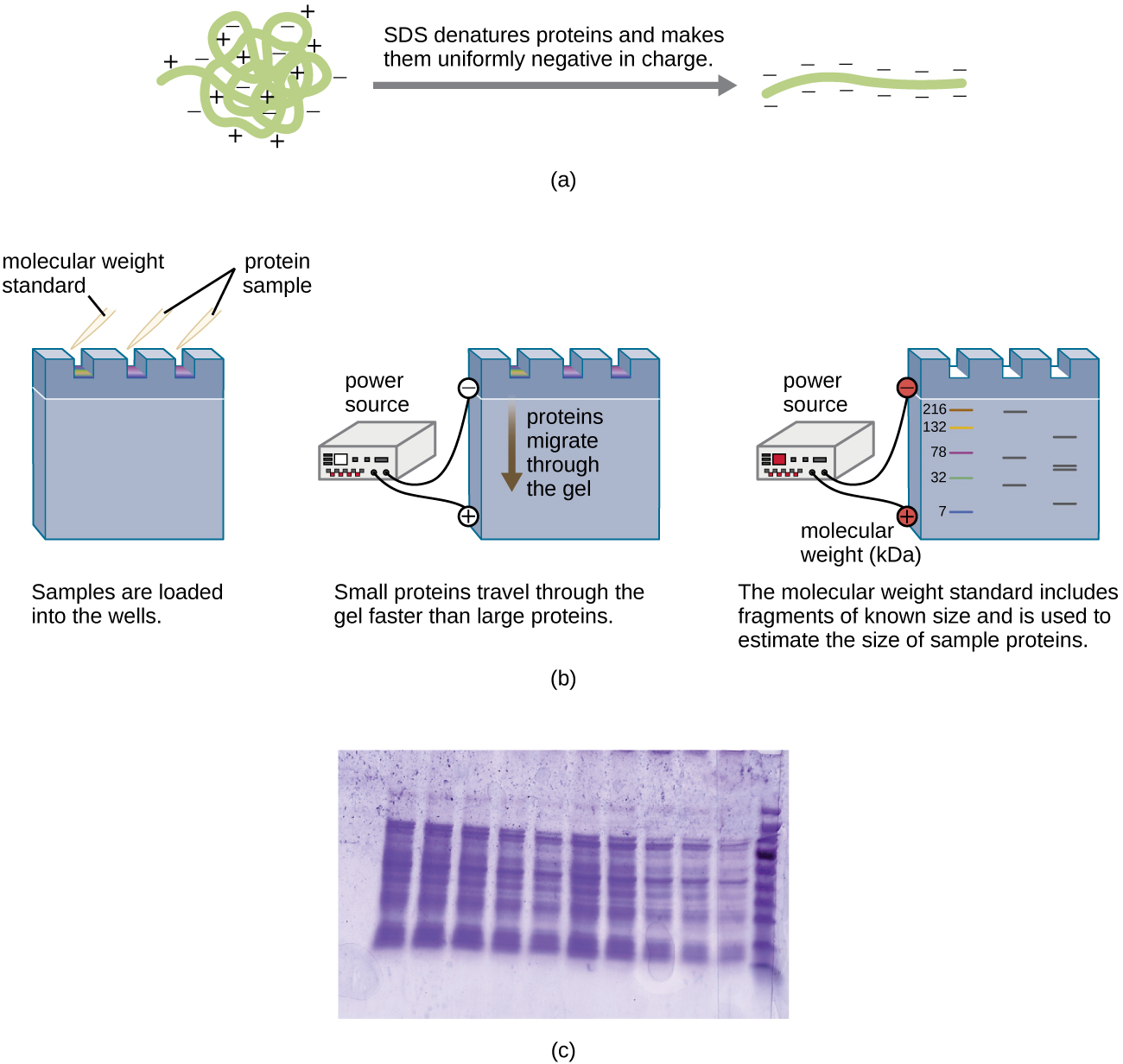
Uma variação da eletroforese em gel, chamada eletroforese em gel de poliacrilamida (PAGE), é comumente usada para separar proteínas. Em PAGE, a matriz de gel é mais fina e composta de poliacrilamida em vez de agarose. Além disso, o PAGE é normalmente executado usando um aparelho de gel vertical (Figura\(\PageIndex{6}\)). Devido às cargas variáveis associadas às cadeias laterais de aminoácidos, o PAGE pode ser usado para separar proteínas intactas com base em suas cargas líquidas. Alternativamente, as proteínas podem ser desnaturadas e revestidas com um detergente de carga negativa chamado dodecil sulfato de sódio (SDS), mascarando as cargas nativas e permitindo a separação com base apenas no tamanho. O PAGE pode ser posteriormente modificado para separar proteínas com base em duas características, como suas cargas em vários pHs e seu tamanho, por meio do uso de PAGE bidimensional. Em qualquer um desses casos, após a eletroforese, as proteínas são visualizadas por meio de coloração, geralmente com coloração azul de Coomassie ou prateada.
Exercício\(\PageIndex{3}\)
Em que base as proteínas são separadas no SDS-PAGE?
Foco clínico: Parte 3
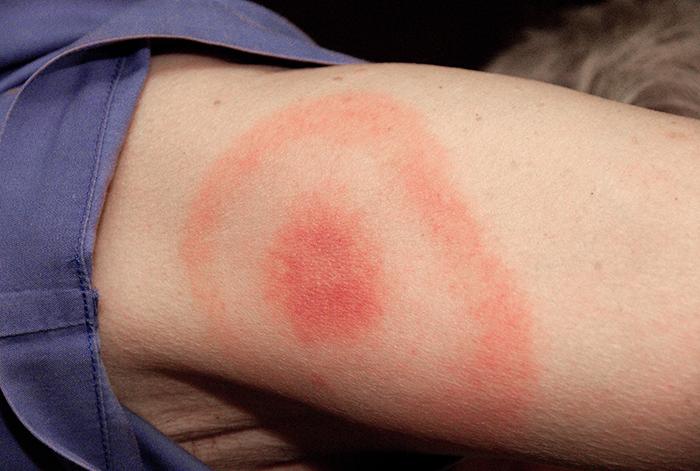
Quando Kayla descreveu seus sintomas, seu médico inicialmente suspeitou de meningite bacteriana, o que é consistente com suas dores de cabeça e rigidez no pescoço. No entanto, ela logo descartou isso como uma possibilidade, porque a meningite geralmente progride mais rapidamente do que a que Kayla estava experimentando. Muitos de seus sintomas ainda eram semelhantes aos da esclerose lateral amiotrófica (ELA) e do lúpus eritematoso sistêmico (LES), e o médico também considerou a doença de Lyme uma possibilidade devido ao tempo que Kayla passa na floresta. Kayla não se lembrava de nenhuma picada recente de carrapato (o meio típico pelo qual a doença de Lyme é transmitida) e ela não tinha a erupção cutânea típica associada à doença de Lyme (Figura\(\PageIndex{7}\)). No entanto, 20 a 30% dos pacientes com doença de Lyme nunca desenvolvem essa erupção cutânea, então o médico não quis descartá-la.
O médico de Kayla solicitou uma ressonância magnética de seu cérebro, um hemograma completo para testar a anemia, exames de sangue avaliando a função hepática e renal e exames adicionais para confirmar ou descartar o LES ou a doença de Lyme. Os resultados do teste foram inconsistentes tanto com o LES quanto com a ELA, e o resultado do teste de busca de anticorpos contra a doença de Lyme foi “equívoco”, ou seja, inconclusivo. Tendo descartado a ELA e o LES, o médico de Kayla decidiu fazer testes adicionais para a doença de Lyme.
Exercício\(\PageIndex{4}\)
- Por que o médico de Kayla ainda suspeitaria da doença de Lyme, mesmo que os resultados do teste não detectassem anticorpos de Lyme no sangue?
- Que tipo de teste molecular pode ser usado para a detecção de anticorpos sanguíneos contra a doença de Lyme?
Métodos de análise de DNA baseados em amplificação
Vários métodos podem ser usados para obter sequências de DNA, que são úteis para estudar organismos causadores de doenças. Com o advento da tecnologia de sequenciamento rápido, nossa base de conhecimento de todos os genomas de organismos patogênicos cresceu fenomenalmente. Começamos com uma descrição da reação em cadeia da polimerase, que não é um método de sequenciamento, mas permitiu que pesquisadores e médicos obtivessem as grandes quantidades de DNA necessárias para o sequenciamento e outros estudos. A reação em cadeia da polimerase elimina a dependência que antes tínhamos das células para fazer várias cópias do DNA, alcançando o mesmo resultado por meio de reações relativamente simples fora da célula.
Reação em cadeia da polimerase (PCR)
A maioria dos métodos de análise de DNA, como digestão enzimática de restrição e eletroforese em gel de agarose, ou sequenciamento de DNA, requerem grandes quantidades de um fragmento de DNA específico. No passado, grandes quantidades de DNA eram produzidas pelo crescimento das células hospedeiras de uma biblioteca genômica. No entanto, as bibliotecas exigem tempo e esforço para serem preparadas e as amostras de DNA de interesse geralmente vêm em quantidades mínimas. A reação em cadeia da polimerase (PCR) permite uma rápida amplificação no número de cópias de sequências de DNA específicas para análise posterior (Figura\(\PageIndex{8}\)). Uma das técnicas mais poderosas em biologia molecular, a PCR foi desenvolvida em 1983 por Kary Mullis enquanto estava na Cetus Corporation. A PCR tem aplicações específicas em laboratórios de pesquisa, forenses e clínicos, incluindo:
- determinando a sequência de nucleotídeos em uma região específica do DNA
- amplificação de uma região alvo do DNA para clonagem em um vetor de plasmídeo
- identificando a fonte de uma amostra de DNA deixada na cena do crime
- analisando amostras para determinar a paternidade
- comparando amostras de DNA antigo com organismos modernos
- determinar a presença de microrganismos difíceis de cultivar ou não cultiváveis em amostras humanas ou ambientais
A PCR é uma técnica de laboratório in vitro que aproveita o processo natural de replicação do DNA. As enzimas de DNA polimerase termoestáveis usadas na PCR são derivadas de procariontes hipertermofílicos. A Taq DNA polimerase, comumente usada em PCR, é derivada da bactéria Thermus aquaticus isolada de uma fonte termal no Parque Nacional de Yellowstone. A replicação do DNA requer o uso de primers para o início da replicação para ter grupos 3'-hidroxila livres disponíveis para a adição de nucleotídeos pela DNA polimerase. No entanto, enquanto os primers compostos de RNA são normalmente usados nas células, os primers de DNA são usados para PCR. Os primers de DNA são preferíveis devido à sua estabilidade, e os primers de DNA com sequências conhecidas direcionadas a uma região específica do DNA podem ser sintetizados quimicamente comercialmente. Esses primers de DNA são funcionalmente semelhantes às sondas de DNA usadas para as várias técnicas de hibridização descritas anteriormente, ligando-se a alvos específicos devido à complementaridade entre a sequência de DNA alvo e o primer.
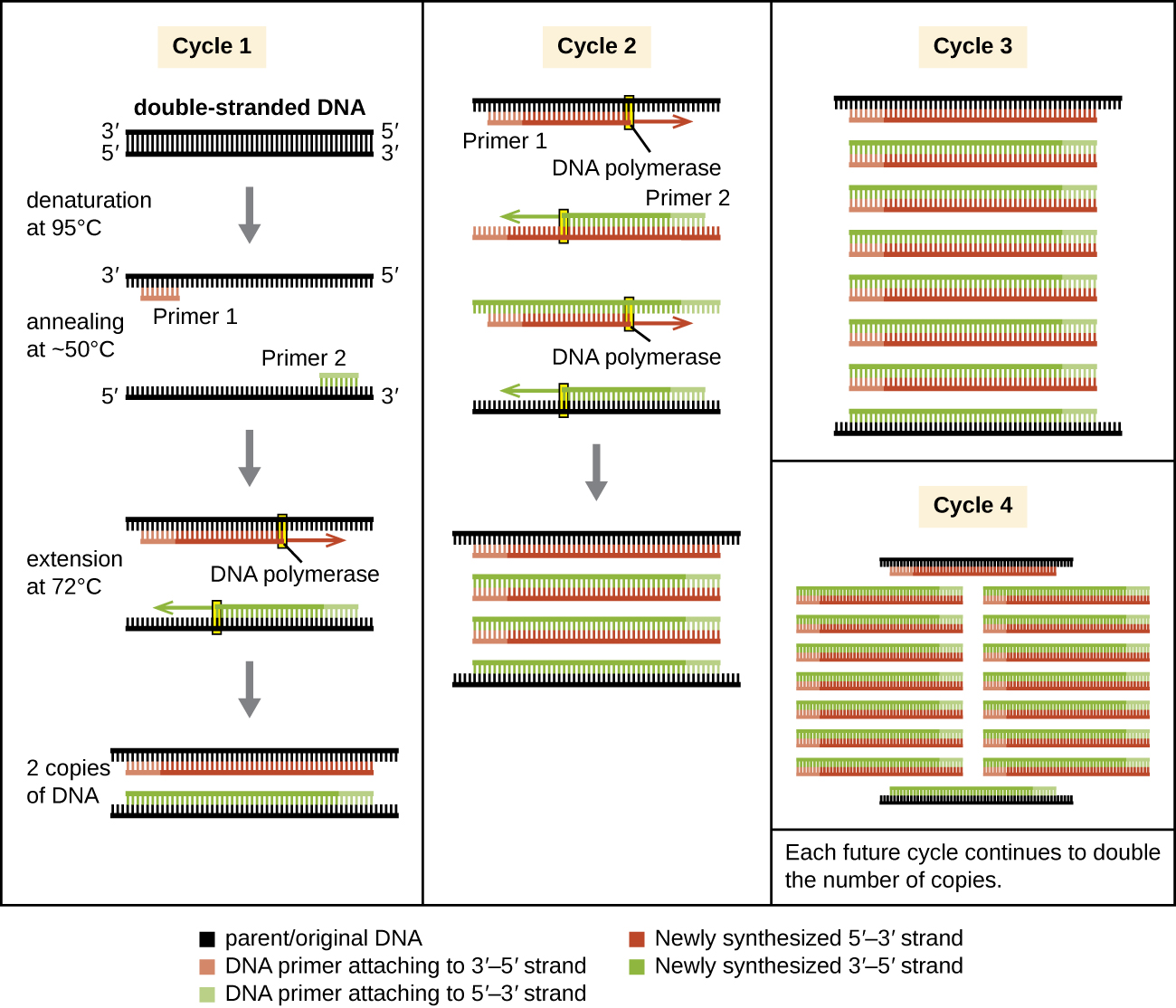
A PCR ocorre em vários ciclos, cada um contendo três etapas: desnaturação, recozimento e extensão. Máquinas chamadas termocicladores são usadas para PCR; essas máquinas podem ser programadas para percorrer automaticamente as temperaturas necessárias em cada etapa (Figura 12.1). Primeiro, o DNA modelo de fita dupla contendo a sequência alvo é desnaturado a aproximadamente 95 °C. A alta temperatura necessária para separar fisicamente (em vez de enzimaticamente) as fitas de DNA é a razão pela qual a DNA polimerase estável ao calor é necessária. Em seguida, a temperatura é reduzida para aproximadamente 50 °C. Isso permite que os primers de DNA complementares às extremidades da sequência alvo se recoloquem (grudem) nas fitas do modelo, com um recozimento primário em cada fita. Finalmente, a temperatura é elevada para 72° C, a temperatura ideal para a atividade da DNA polimerase estável ao calor, permitindo a adição de nucleotídeos ao primer usando o alvo de fita simples como modelo. Cada ciclo dobra o número de cópias de DNA alvo de fita dupla. Normalmente, os protocolos de PCR incluem de 25 a 40 ciclos, permitindo a amplificação de uma única sequência alvo em dezenas de milhões para mais de um trilhão.
A replicação natural do DNA é projetada para copiar todo o genoma e se inicia em um ou mais locais de origem. Os primers são construídos durante a replicação, não antes, e não consistem em algumas sequências específicas. A PCR tem como alvo regiões específicas de uma amostra de DNA usando primers específicos da sequência. Nos últimos anos, uma variedade de métodos de amplificação isotérmica por PCR que contornam a necessidade de ciclagem térmica foram desenvolvidos, aproveitando as proteínas acessórias que auxiliam no processo de replicação do DNA. À medida que o desenvolvimento desses métodos continua e seu uso se torna mais difundido em laboratórios de pesquisa, forenses e clínicos, os termocicladores podem se tornar obsoletos.
Aprofunde sua compreensão da reação em cadeia da polimerase vendo esta animação e trabalhando em um exercício interativo.
Variações de PCR
Várias modificações posteriores na PCR aumentam ainda mais a utilidade dessa técnica. A PCR de transcriptase reversa (RT-PCR) é usada para obter cópias de DNA de uma molécula específica de mRNA. A RT-PCR começa com o uso da enzima transcriptase reversa para converter moléculas de mRNA em cDNA. Esse cDNA é então usado como um modelo para a amplificação tradicional de PCR. A RT-PCR pode detectar se um gene específico foi expresso em uma amostra. Outra aplicação recente da PCR é a PCR em tempo real, também conhecida como PCR quantitativa (qPCR). Os protocolos padrão de PCR e RT-PCR não são quantitativos porque qualquer um dos reagentes pode se tornar limitante antes que todos os ciclos do protocolo sejam concluídos e as amostras sejam analisadas apenas no final. Como não é possível determinar quando, no protocolo de PCR ou RT-PCR, um determinado reagente se tornou limitante, não é possível saber quantos ciclos foram concluídos antes desse ponto e, portanto, não é possível determinar quantas moléculas modelo originais estavam presentes na amostra no início de PCR. No qPCR, no entanto, o uso de fluorescência permite monitorar o aumento em um molde de fita dupla durante uma reação de PCR à medida que ela ocorre. Esses dados cinéticos podem então ser usados para quantificar a quantidade da sequência alvo original. O uso do qPCR nos últimos anos expandiu ainda mais as capacidades da PCR, permitindo que os pesquisadores determinem o número de cópias de DNA e, às vezes, organismos, presentes em uma amostra. Em ambientes clínicos, o qRT-PCR é usado para determinar a carga viral em pacientes HIV-positivos para avaliar a eficácia de sua terapia.
Sequenciamento de DNA
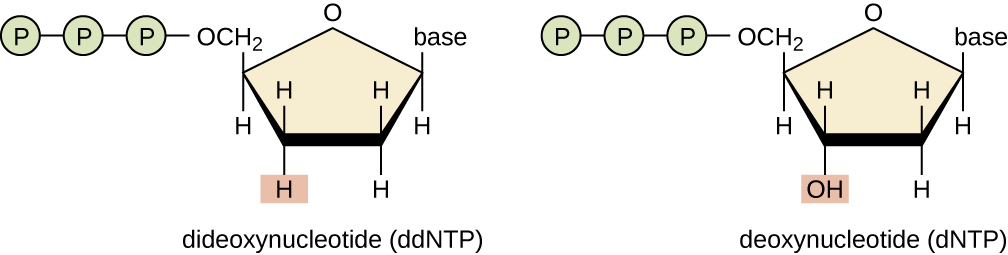
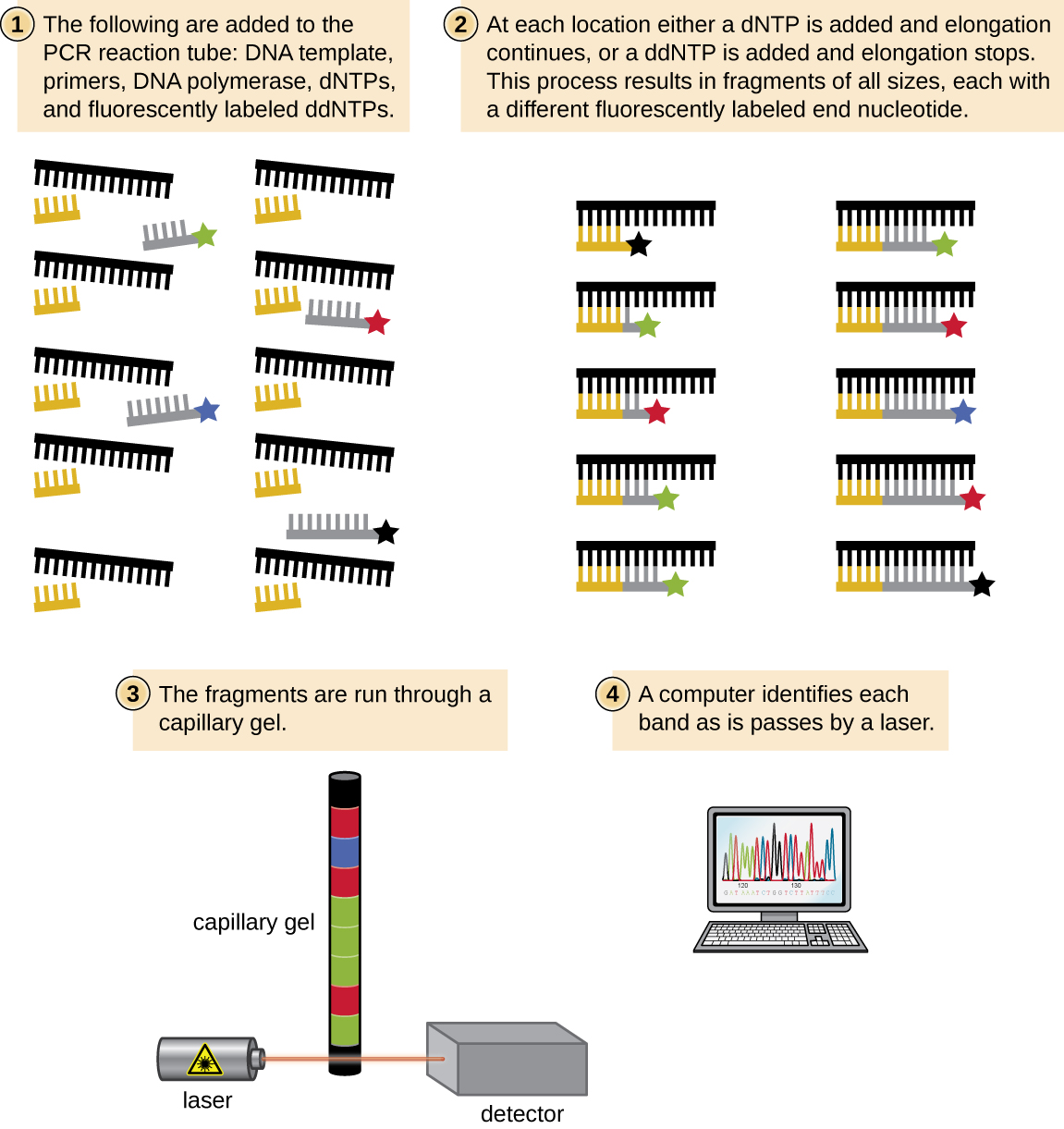
Uma técnica básica de sequenciamento é o método de terminação em cadeia, também conhecido como método dideoxi ou método de sequenciamento de DNA Sanger, desenvolvido por Frederick Sanger em 1972. O método de terminação em cadeia envolve a replicação do DNA de um modelo de fita simples com o uso de um primer de DNA para iniciar a síntese de uma fita complementar, a DNA polimerase, uma mistura dos quatro monômeros regulares de desoxinucleotídeos (dNTP) e uma pequena proporção de didesoxinucleotídeos (DDNTPs), cada um marcado com um farol molecular. Os DDNTPs são monômeros sem um grupo hidroxila (—OH) no local em que outro nucleotídeo geralmente se liga para formar uma cadeia (Figura\(\PageIndex{9}\)). Toda vez que um dDNTP é incorporado aleatoriamente à fita complementar em crescimento, ele encerra o processo de replicação do DNA dessa fita específica. Isso resulta em várias fitas curtas de DNA replicado, cada uma terminada em um ponto diferente durante a replicação. Quando a mistura de reação é submetida à eletroforese em gel, as múltiplas fitas de DNA recém-replicadas formam uma escada de tamanhos diferentes. Como os dDNTPs são rotulados, cada banda no gel reflete o tamanho da fita de DNA quando o dDNTP encerrou a reação.
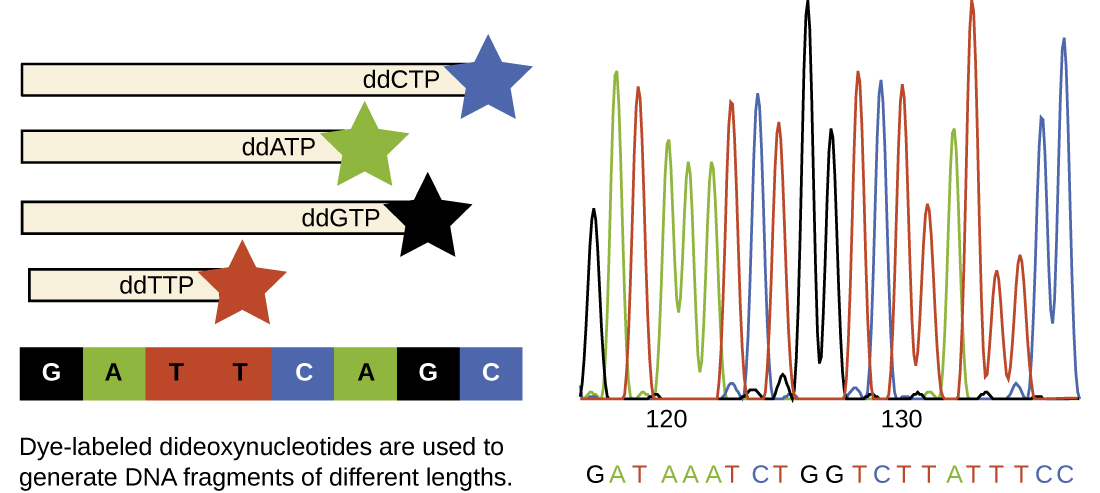
Na época de Sanger, quatro reações foram configuradas para cada molécula de DNA sendo sequenciada, cada reação contendo apenas um dos quatro possíveis DDNTPs. Cada dDNTP foi marcado com uma molécula radioativa de fósforo. Os produtos das quatro reações foram então executados em faixas separadas lado a lado em géis PAGE longos e estreitos, e as faixas de comprimentos variados foram detectadas por autorradiografia. Hoje, esse processo foi simplificado com o uso de DDNTPs, cada um marcado com um corante fluorescente ou fluorocromo de cor diferente (Figura\(\PageIndex{10}\)), em uma reação de sequenciamento contendo todos os quatro DDNTPs possíveis para cada molécula de DNA sendo sequenciada (Figura\(\PageIndex{11}\)). Esses fluorocromos são detectados por espectroscopia de fluorescência. Determinar a cor de fluorescência de cada banda à medida que ela passa pelo detector produz a sequência de nucleotídeos da fita modelo.
Desde 2005, as técnicas de sequenciamento automatizado usadas pelos laboratórios estão sob a égide do sequenciamento de próxima geração, que é um grupo de técnicas automatizadas usadas para sequenciamento rápido de DNA. Esses métodos revolucionaram o campo da genética molecular porque os sequenciadores de baixo custo podem gerar sequências de centenas de milhares ou milhões de fragmentos curtos (25 a 600 pares de bases) em apenas um dia. Embora várias variantes das tecnologias de sequenciamento de próxima geração sejam feitas por empresas diferentes (por exemplo, o pirosequenciamento da 454 Life Sciences e a tecnologia Solexa da Illumina), todas elas permitem que milhões de bases sejam sequenciadas rapidamente, tornando o sequenciamento de genomas inteiros relativamente fácil e barato, e corriqueiro. No sequenciamento 454 (pirosequenciamento), por exemplo, uma amostra de DNA é fragmentada em fragmentos de fita única de 400—600 pb, modificados com a adição de adaptadores de DNA nas duas extremidades de cada fragmento. Cada fragmento de DNA é então imobilizado em um cordão e amplificado por PCR, usando primers projetados para recozer os adaptadores, criando um cordão contendo muitas cópias desse fragmento de DNA. Cada grânulo é então colocado em um poço separado contendo enzimas de sequenciamento. Ao poço, cada um dos quatro nucleotídeos é adicionado um após o outro; quando cada um é incorporado, o pirofosfato é liberado como subproduto da polimerização, emitindo um pequeno flash de luz registrado por um detector. Isso fornece a ordem dos nucleotídeos incorporados à medida que uma nova fita de DNA é feita e é um exemplo de sequenciamento de síntese. Os sequenciadores de próxima geração usam software sofisticado para superar o complicado processo de colocar todos os fragmentos em ordem. No geral, essas tecnologias continuam avançando rapidamente, diminuindo o custo do sequenciamento e aumentando rapidamente a disponibilidade de dados de sequência de uma ampla variedade de organismos.
O National Center for Biotechnology Information abriga um banco de dados de sequências genéticas amplamente usado chamado GenBank, onde pesquisadores depositam informações genéticas para uso público. Após a publicação dos dados da sequência, os pesquisadores os enviam para o GenBank, dando a outros pesquisadores acesso às informações. A colaboração permite que os pesquisadores comparem informações de sequências de amostras recém-descobertas ou desconhecidas com a vasta gama de dados de sequência que já existe.
Veja uma animação sobre o sequenciamento 454 para aprofundar sua compreensão desse método.
Usando um NAAT para diagnosticar uma infecção por C. difficile
Javier, um paciente de 80 anos com histórico de doença cardíaca, voltou recentemente para casa do hospital após passar por um procedimento de angioplastia para inserir um stent em uma artéria cardíaca. Para minimizar a possibilidade de infecção, Javier recebeu antibióticos intravenosos de amplo espectro durante e logo após o procedimento. Ele recebeu alta quatro dias após o procedimento, mas uma semana depois, começou a sentir cólicas abdominais leves e diarreia aquosa várias vezes ao dia. Ele perdeu o apetite, ficou gravemente desidratado e desenvolveu febre. Ele também notou sangue nas fezes. A esposa de Javier ligou para o médico, que a instruiu a levá-lo imediatamente ao pronto-socorro.
A equipe do hospital fez vários testes e descobriu que os níveis de creatinina nos rins de Javier estavam elevados em comparação com os níveis em seu sangue, indicando que seus rins não estavam funcionando bem. Os sintomas de Javier sugeriram uma possível infecção por Clostridium difficile, uma bactéria resistente a muitos antibióticos. O hospital coletou e cultivou uma amostra de fezes para verificar a produção das toxinas A e B por C. difficile, mas os resultados foram negativos. No entanto, os resultados negativos não foram suficientes para descartar uma infecção por C. difficile porque o cultivo de C. difficile e a detecção de suas toxinas características podem ser difíceis, particularmente em alguns tipos de amostras. Por segurança, eles realizaram um teste diagnóstico de amplificação de ácido nucléico (NAAT). Atualmente, os NAATs são o padrão-ouro do diagnosticador clínico para detectar o material genético de um patógeno. No caso de Javier, o qPCR foi usado para procurar o gene que codifica a toxina B do C. difficile (TcDB). Quando a análise de qPCR deu positivo, o médico assistente concluiu que Javier estava realmente sofrendo de uma infecção por C. difficile e imediatamente prescreveu o antibiótico vancomicina, para ser administrado por via intravenosa. O antibiótico eliminou a infecção e Javier se recuperou completamente.
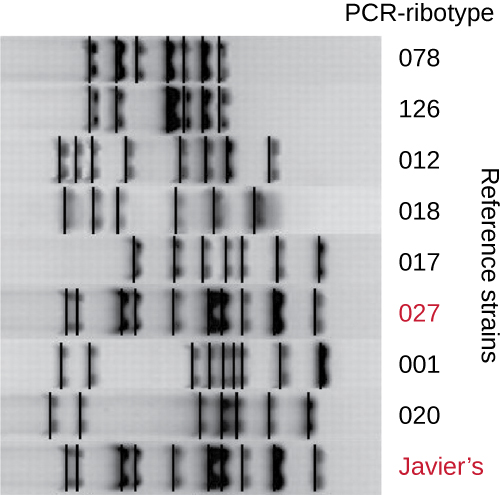
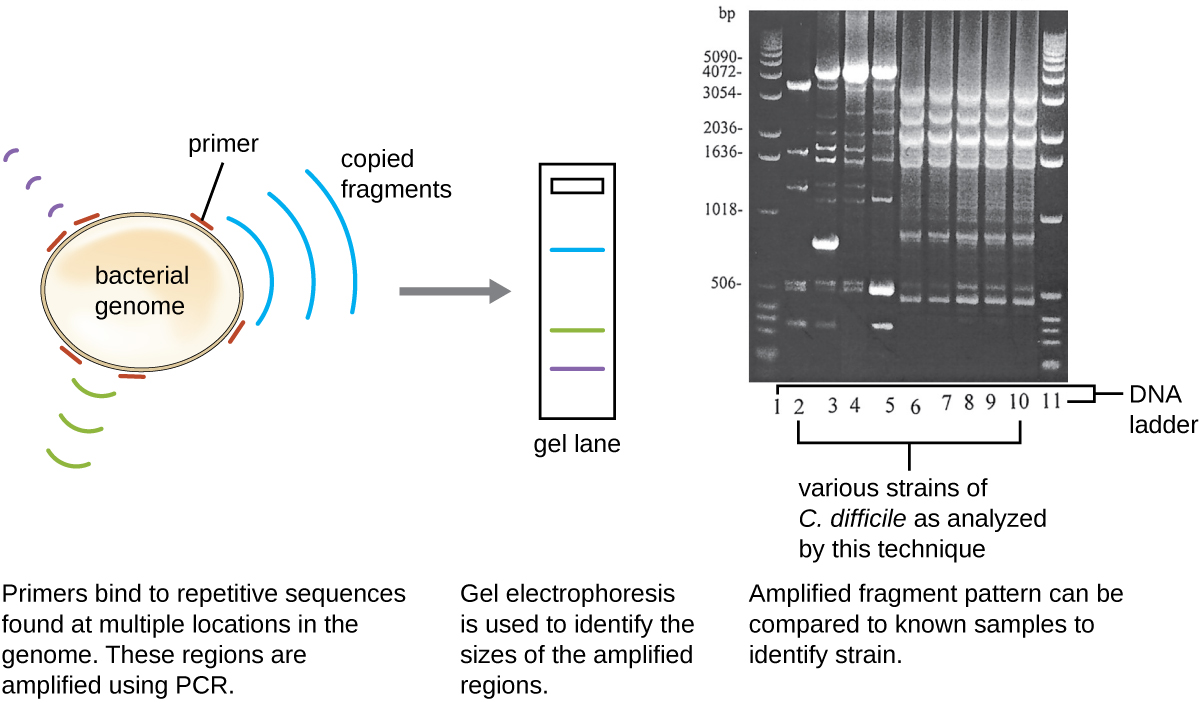
Como as infecções por C. difficile estavam se espalhando pela comunidade de Javier, sua amostra foi posteriormente analisada para verificar se a cepa específica de C. difficile poderia ser identificada. A amostra de fezes de Javier foi submetida à ribotipagem e análise de PCR baseada em sequência repetitiva (Rep-PCR). Na ribotipagem, uma pequena sequência de DNA entre os genes 16S rRNA e 23S rRNA é amplificada e submetida à digestão por restrição (Figura\(\PageIndex{12}\)). Essa sequência varia entre as cepas de C. difficile, portanto, as enzimas de restrição cortarão em lugares diferentes. Em Rep-PCR, primers de DNA projetados para se ligar a sequências curtas comumente encontradas repetidas dentro do genoma de C. difficile foram usados para PCR. Após a digestão por restrição, a eletroforese em gel de agarose foi realizada em ambos os tipos de análise para examinar os padrões de bandagem resultantes de cada procedimento (Figura\(\PageIndex{13}\)). O Rep-PCR pode ser usado para subtipar ainda mais vários ribotipos, aumentando a resolução para detectar diferenças entre cepas. Verificou-se que o ribotipo da cepa que infecta Javier é o ribotipo 27, uma cepa conhecida por sua maior virulência, resistência a antibióticos e aumento da prevalência nos Estados Unidos, Canadá, Japão e Europa. 1
Exercício\(\PageIndex{5}\)
- Como os padrões de bandas diferem entre as cepas de C. difficile?
- Por que você acha que os testes de laboratório não conseguiram detectar diretamente a produção de toxinas?
Exercício\(\PageIndex{6}\)
- Como a PCR é semelhante ao processo natural de replicação do DNA nas células? Como isso é diferente?
- Compare o RT-PCR e o qPCR em termos de suas respectivas finalidades.
- No sequenciamento de terminação em cadeia, como a identidade de cada nucleotídeo em uma sequência é determinada?
Conceitos principais e resumo
- Encontrar um gene de interesse em uma amostra requer o uso de uma sonda de DNA de fita simples marcada com um farol molecular (normalmente radioatividade ou fluorescência) que pode hibridizar com um ácido nucléico de fita simples complementar na amostra.
- A eletroforese em gel de agarose permite a separação de moléculas de DNA com base no tamanho.
- A análise do polimorfismo de comprimento de fragmento de restrição (RFLP) permite a visualização por eletroforese em gel de agarose de variantes distintas de uma sequência de DNA causadas por diferenças nos locais de restrição.
- A análise de Southern blot permite que os pesquisadores encontrem uma sequência de DNA específica em uma amostra, enquanto a análise de Northern blot permite que os pesquisadores detectem uma sequência específica de mRNA expressa em uma amostra.
- A tecnologia de microarray é uma técnica de hibridização de ácido nucléico que permite o exame de muitos milhares de genes ao mesmo tempo para encontrar diferenças nos genes ou padrões de expressão gênica entre duas amostras de DNA genômico ou cDNA,
- A eletroforese em gel de poliacrilamida (PAGE) permite a separação de proteínas por tamanho, especialmente se as cargas proteicas nativas forem mascaradas por meio de pré-tratamento com SDS.
- A reação em cadeia da polimerase permite a rápida amplificação de uma sequência específica de DNA. Variações de PCR podem ser usadas para detectar a expressão de mRNA (PCR da transcriptase reversa) ou para quantificar uma sequência específica na amostra original (PCR em tempo real).
- Embora o desenvolvimento do sequenciamento de DNA Sanger tenha sido revolucionário, os avanços no sequenciamento de próxima geração permitem o sequenciamento rápido e barato dos genomas de muitos organismos, acelerando o volume de novos dados de sequência.
Notas de pé
- 1 Patrizia Spigaglia, Fabrizio Barbanti, Anna Maria Dionisi e Paola Mastrantonio. “Isolados de Clostridium difficile resistentes às fluoroquinolonas na Itália: surgimento do ribotipo 018 de PCR.” Jornal de Microbiologia Clínica 48 nº 8 (2010): 2892—2896.