
14.3 : Médicaments ciblant d'autres microorganismes
- Page ID
- 187923

Objectifs d'apprentissage
- Décrire les mécanismes d'action associés aux médicaments qui inhibent la biosynthèse des parois cellulaires, la synthèse des protéines, la fonction membranaire, la synthèse des acides nucléiques et les voies métaboliques.
L'une des qualités importantes d'un médicament antimicrobien est sa toxicité sélective, c'est-à-dire qu'il tue ou inhibe de manière sélective la croissance des cibles microbiennes tout en causant des dommages minimes ou nuls à l'hôte. La plupart des médicaments antimicrobiens actuellement utilisés en clinique sont antibactériens, car les cellules procaryotes fournissent une plus grande variété de cibles uniques de toxicité sélective, par rapport aux champignons, aux parasites et aux virus. Chaque classe de médicaments antibactériens possède un mode d'action unique (la manière dont un médicament affecte les microbes au niveau cellulaire), qui est résumé dans la figure\(\PageIndex{1}\) et le tableau\(\PageIndex{1}\).
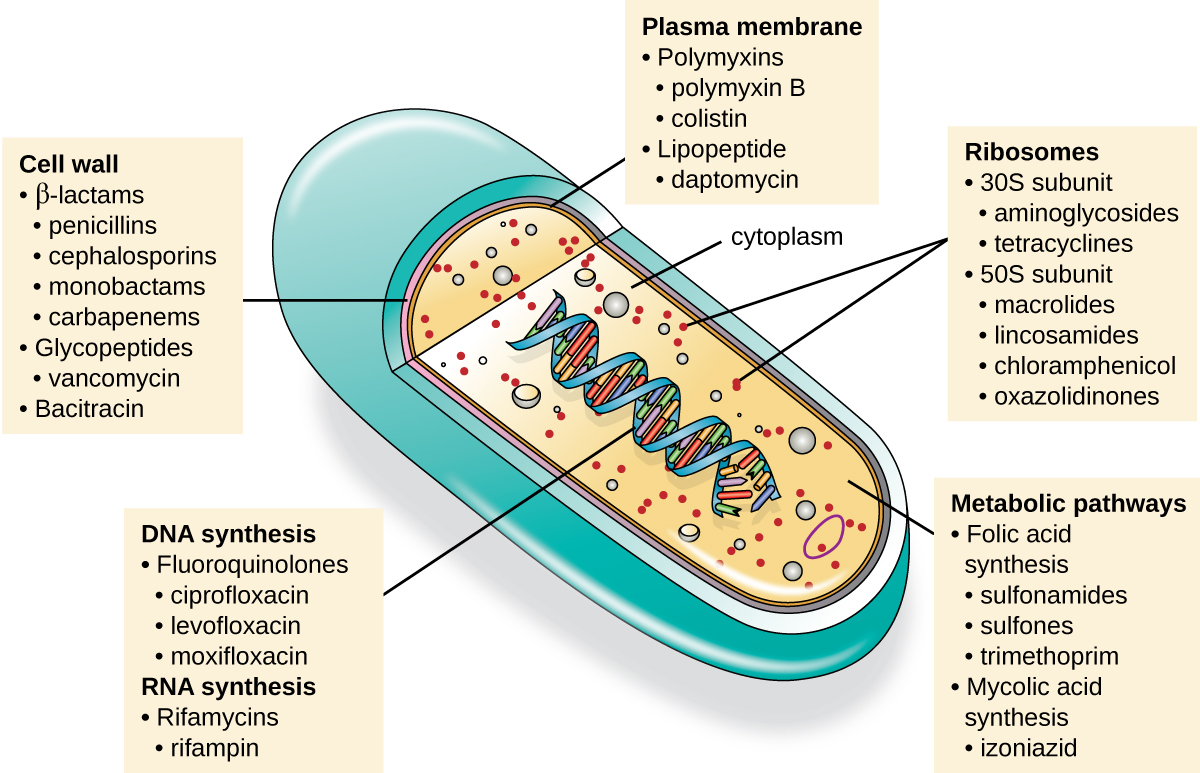
| Mode d'action | Cible | Classe de médicaments |
|---|---|---|
| Inhiber la biosynthèse des parois | Protéines liant la pénicilline | β-lactames : pénicillines, céphalosporines, monobactames, carbapénèmes |
| Sous-unités du peptidoglycane | Glycopeptides | |
| Transport des sous-unités du peptidoglycane | Bacitracine | |
| Inhiber la biosynthèse des protéines | Sous-unité ribosomale 30S | Aminoglycosides, tétracyclines |
| Sous-unité ribosomale 50S | Macrolides, lincosamides, chloramphénicol, oxazolidinones | |
| Interrompre les membranes | Lipopolysaccharide, membranes interne et externe | Polymyxine B, colistine, daptomycine |
| Inhiber la synthèse des acides | ARN | Rifamycine |
| ADN | Fluoroquinolones | |
| Antimétabolites | enzyme de synthèse de l'acide folique | Sulfamides, triméthoprime |
| enzyme de synthèse de l'acide mycolique | Hydrazide d'acide isonicotinique | |
| Inhibiteur mycobactérien de l'adénosine triphosphate (ATP) synthase | ATP synthase mycobactérienne | Diarylquinoléine |
Inhibiteurs de la biosynthèse des parois cellulaires
Plusieurs classes différentes d'antibactériens bloquent les étapes de la biosynthèse du peptidoglycane, rendant les cellules plus sensibles à la lyse osmotique (Tableau\(\PageIndex{2}\)). Par conséquent, les antibactériens qui ciblent la biosynthèse des parois cellulaires ont une action bactéricide. Comme les cellules humaines ne produisent pas de peptidoglycane, ce mode d'action est un excellent exemple de toxicité sélective.
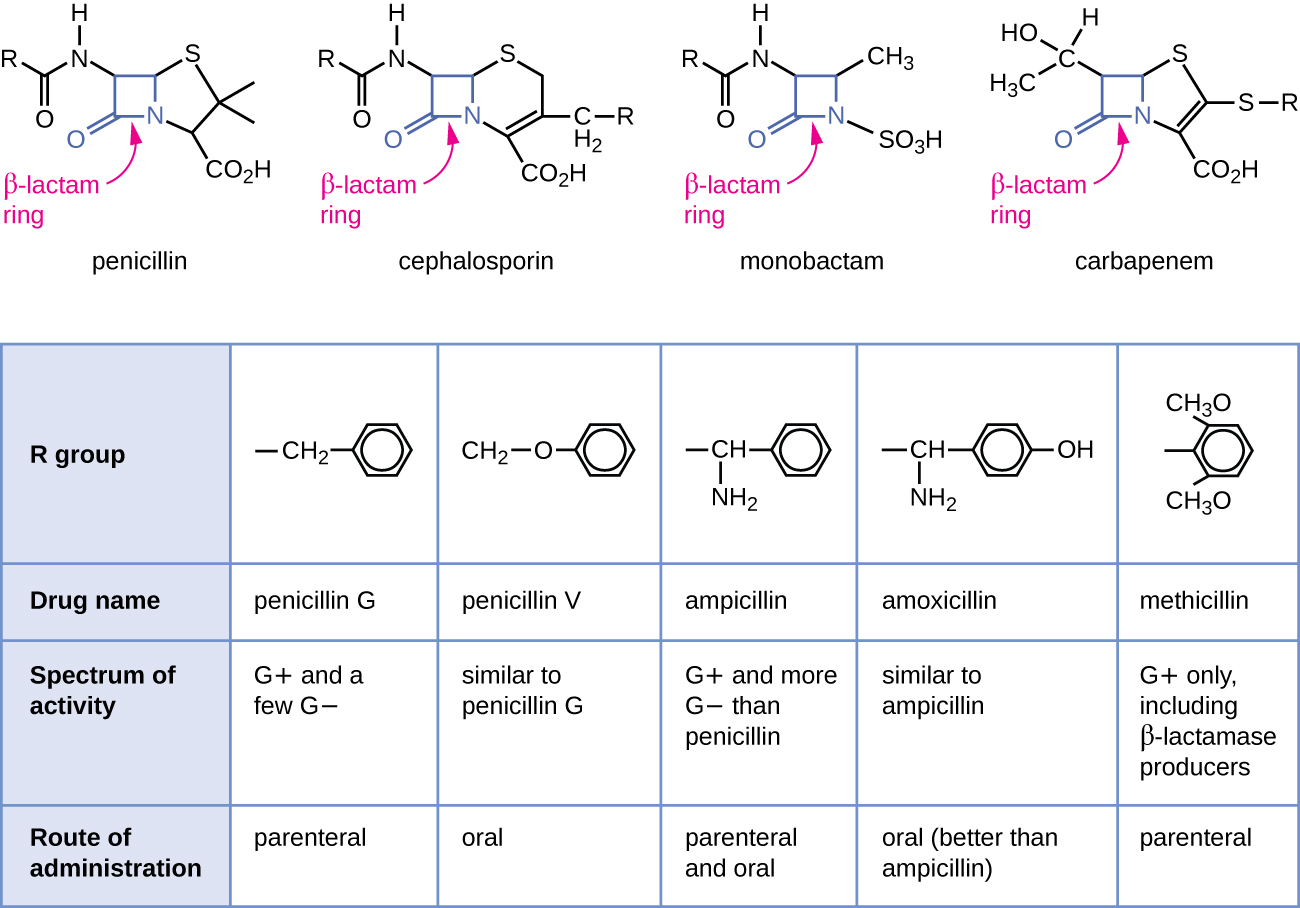
La pénicilline, le premier antibiotique découvert, est l'un des nombreux antibactériens de la classe des β-lactamines. Ce groupe de composés comprend les pénicillines, les céphalosporines, les monobactames et les carbapénèmes, et se caractérise par la présence d'un cycle β-lactame présent dans la structure centrale de la molécule médicamenteuse (Figure\(\PageIndex{2}\)). Les antibactériens à base de β-lactamines bloquent la réticulation des chaînes peptidiques lors de la biosynthèse du nouveau peptidoglycane dans la paroi cellulaire bactérienne. Ils sont capables de bloquer ce processus parce que la structure du β-lactame est similaire à celle du composant de la sous-unité du peptidoglycane reconnu par l'enzyme transpeptidase réticulante, également connue sous le nom de protéine liant la pénicilline (PBP). Bien que le cycle β-lactame doive rester inchangé pour que ces médicaments conservent leur activité antibactérienne, des modifications chimiques stratégiques apportées aux groupes R ont permis le développement d'une grande variété de médicaments semi-synthétiques à base de β-lactame présentant une activité accrue, un spectre d'activité élargi et des demi-vies plus longues pour une meilleure dosage, entre autres caractéristiques.
La pénicilline G et la pénicilline V sont des antibiotiques naturels issus de champignons et sont principalement actives contre les bactéries pathogènes à Gram positif et contre quelques agents pathogènes bactériens à Gram négatif tels que Pasteurella multocida. La figure\(\PageIndex{2}\) résume le développement semi-synthétique de certaines pénicillines. L'ajout d'un groupe amino (-NH 2) à la pénicilline G a créé les aminopénicillines (c'est-à-dire l'ampicilline et l'amoxicilline) qui ont un spectre d'activité accru contre un plus grand nombre d'agents pathogènes à Gram négatif. De plus, l'ajout d'un groupe hydroxyle (-OH) à l'amoxicilline a augmenté la stabilité acide, ce qui permet une meilleure absorption orale. La méthicilline est une pénicilline semi-synthétique qui a été développée pour lutter contre la propagation d'enzymes (pénicillinases) qui inactivaient les autres pénicillines. Le remplacement du groupe R de la pénicilline G par le groupe diméthoxyphényle, plus volumineux, a protégé le cycle β-lactame contre la destruction enzymatique par les pénicillinases, nous donnant ainsi la première pénicilline résistante à la pénicillinase.
Comme les pénicillines, les céphalosporines contiennent un cycle β-lactame (Figure\(\PageIndex{2}\)) et bloquent l'activité transpeptidase des protéines liant les pénicillines. Cependant, le cycle β-lactame des céphalosporines est fusionné à un cycle à six chaînons, plutôt qu'au cycle à cinq chaînons présent dans les pénicillines. Cette différence chimique confère aux céphalosporines une résistance accrue à l'inactivation enzymatique par les β-lactamases. Le médicament céphalosporine C a été isolé à l'origine du champignon Cephalosporium acremonium dans les années 1950 et possède un spectre d'activité similaire à celui de la pénicilline contre les bactéries gram-positives, mais il est actif contre un plus grand nombre de bactéries gram-négatives que la pénicilline. Une autre différence structurelle importante est que la céphalosporine C possède deux groupes R, contre un seul groupe R pour la pénicilline, ce qui permet une plus grande diversité dans les altérations chimiques et le développement de céphalosporines semi-synthétiques. La famille des céphalosporines semi-synthétiques est beaucoup plus vaste que celle des pénicillines, et ces médicaments ont été classés en générations en fonction principalement de leur spectre d'activité, leur spectre allant des céphalosporines de première génération à spectre étroit aux céphalosporines à large spectre de quatrième génération céphalosporines. Une nouvelle céphalosporine de cinquième génération a été développée, active contre le Staphylococcus aureus résistant à la méthicilline (SARM).
Les carbapénèmes et les monobactames ont également un cycle β-lactame dans leur structure centrale et ils inhibent l'activité transpeptidase des protéines liant la pénicilline. Le seul monobactam utilisé cliniquement est l'aztréonam. C'est un antibactérien à spectre étroit qui agit uniquement contre les bactéries gram-négatives. En revanche, la famille des carbapénèmes comprend divers médicaments semi-synthétiques (imipénème, méropénem et doripénème) qui fournissent une activité à très large spectre contre les bactéries pathogènes gram-positives et gram-négatives.
Le médicament vancomycine, membre d'une classe de composés appelés glycopeptides, a été découvert dans les années 1950 en tant qu'antibiotique naturel issu de l'actinomycète Amycolatopsis orientalis. Comme les β-lactamines, la vancomycine inhibe la biosynthèse des parois cellulaires et est bactéricide. Cependant, contrairement aux β-lactamines, la structure de la vancomycine n'est pas similaire à celle des sous-unités peptidoglycanes de la paroi cellulaire et n'inactive pas directement les protéines liant les pénicillines. La vancomycine est plutôt une très grosse molécule complexe qui se lie à l'extrémité de la chaîne peptidique des précurseurs de la paroi cellulaire, créant ainsi un blocage structurel qui empêche l'incorporation des sous-unités de la paroi cellulaire dans le squelette en croissance de la N-acétylglucosamine et de l'acide N-acétylmuramique (NAM-NAG) du peptidoglycane structure (transglycosylation). La vancomycine bloque également structurellement la transpeptidation. La vancomycine est bactéricide contre les bactéries pathogènes gram-positives, mais elle n'est pas active contre les bactéries gram-négatives en raison de son incapacité à pénétrer la membrane externe protectrice.
Le médicament bacitracine est constitué d'un groupe d'antibiotiques peptidiques structurellement similaires isolés à l'origine à partir de Bacillus subtilis. La bacitracine bloque l'activité d'une molécule membranaire cellulaire spécifique responsable du mouvement des précurseurs des peptidoglycanes du cytoplasme vers l'extérieur de la cellule, empêchant ainsi leur incorporation dans la paroi cellulaire. La bacitracine est efficace contre un large éventail de bactéries, y compris les organismes à Gram positif présents sur la peau, tels que les staphylocoques et les streptocoques. Bien qu'elle puisse être administrée par voie orale ou intramusculaire dans certaines circonstances, la bacitracine s'est révélée néphrotoxique (endommageant les reins). Par conséquent, il est plus souvent associé à la néomycine et à la polymyxine dans des pommades topiques telles que la néosporine.
| Mécanisme d'action | Classe de médicaments | Médicaments spécifiques | Naturel ou semi-synthétique | Spectre d'activité |
|---|---|---|---|---|
| Interagissez directement avec les PBP et inhibez l'activité de la transpeptidase | Pénicillines | Pénicilline G, pénicilline V | Naturel | Spectre étroit contre les bactéries gram-positives et quelques bactéries gram-négatives |
| Ampicilline, amoxicilline | Semisynthétique | Spectre étroit contre les bactéries gram-positives mais spectre gram-négatif accru | ||
| Méthicilline | Semisynthétique | Spectre étroit contre les bactéries gram-positives uniquement, y compris les souches produisant de la pénicillinase | ||
| Céphalosporines | Céphalosporine C | Naturel | Spectre étroit similaire à la pénicilline mais avec un spectre gram-négatif accru | |
| Céphalosporines de première génération | Semisynthétique | Spectre étroit similaire à la céphalosporine C | ||
| Céphalosporines de deuxième génération | Semisynthétique | Spectre étroit mais avec un spectre gram-négatif accru par rapport à la première génération | ||
| Céphalosporines de troisième et quatrième générations | Semisynthétique | Large spectre contre les bactéries gram-positives et gram-négatives, y compris certains producteurs de β-lactamases | ||
| Céphalosporines de cinquième génération | Semisynthétique | Large spectre contre les bactéries gram-positives et gram-négatives, y compris le SARM | ||
| Monobactames | Aztréonam | Semisynthétique | Spectre étroit contre les bactéries gram-négatives, y compris certains producteurs de β-lactamase | |
| Carbapénèmes | Imipénème, méropénem, doripénème | Semisynthétique | Le plus large spectre de β-lactamines contre les bactéries gram-positives et gram-négatives, y compris de nombreux producteurs de β-lactamases | |
| Grandes molécules qui se lient à la chaîne peptidique des sous-unités du peptidoglycane, bloquant ainsi la transglycosylation et la transpeptidation | Glycopeptides | Vancomycine | Naturel | Spectre étroit contre les bactéries gram-positives uniquement, y compris les souches multirésistantes |
| Bloquer le transport des sous-unités du peptidoglycane à travers la membrane cytoplasmique | Bacitracine | Bacitracine | Naturel | Large spectre contre les bactéries gram-positives et gram-négatives |
Exercice\(\PageIndex{1}\)
Décrire le mode d'action des β-lactamines.
Inhibiteurs de biosynthèse des protéines
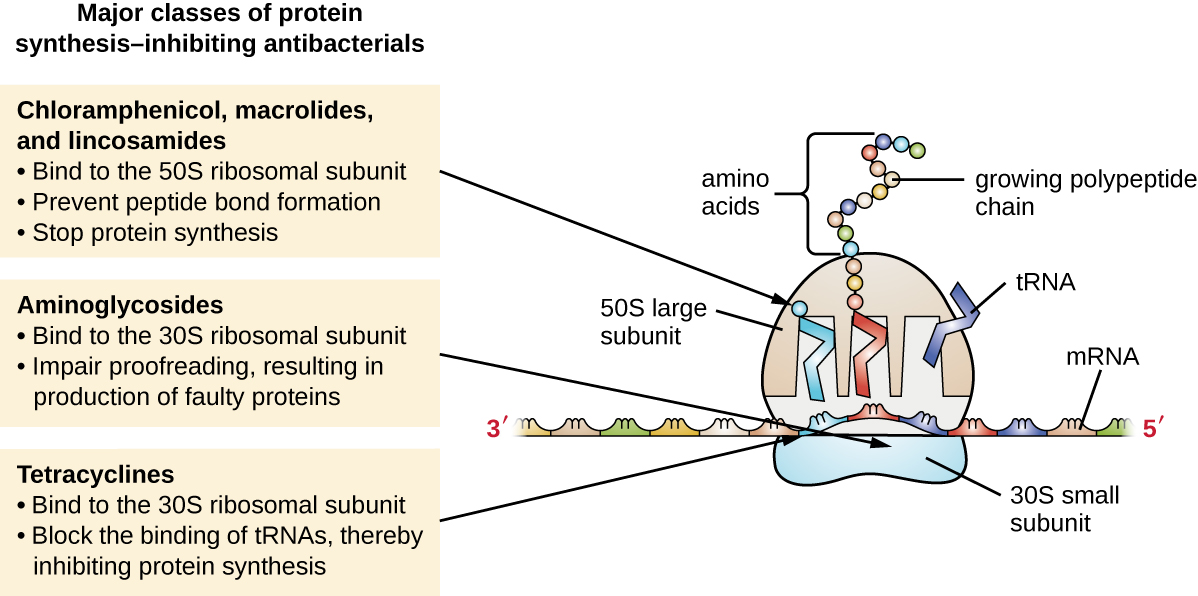
Les ribosomes cytoplasmiques présents dans les cellules animales (80S) sont structurellement distincts de ceux trouvés dans les cellules bactériennes (70S), faisant de la biosynthèse des protéines une bonne cible sélective pour les médicaments antibactériens. Plusieurs types d'inhibiteurs de la biosynthèse des protéines sont discutés dans cette section et sont résumés dans la Figure\(\PageIndex{3}\).
Inhibiteurs de synthèse des protéines qui lient la sous-unité 30S
Les aminoglycosides sont de gros médicaments antibactériens hautement polaires qui se lient à la sous-unité 30S des ribosomes bactériens, altérant ainsi la capacité de relecture du complexe ribosomal. Cette altération provoque des discordances entre les codons et les anticodons, ce qui entraîne la production de protéines contenant des acides aminés incorrects et de protéines raccourcies qui s'insèrent dans la membrane cytoplasmique. La rupture de la membrane cytoplasmique par les protéines défectueuses tue les cellules bactériennes. Les aminoglycosides, qui comprennent des médicaments tels que la streptomycine, la gentamicine, la néomycine et la kanamycine, sont de puissants antibactériens à large spectre. Cependant, il a été démontré que les aminoglycosides sont néphrotoxiques (endommagement des reins), neurotoxiques (endommagement du système nerveux) et ototoxiques (endommagement de l'oreille).
Les tétracyclines constituent une autre classe de composés antibactériens qui se lient à la sous-unité 30S. Contrairement aux aminoglycosides, ces médicaments sont bactériostatiques et inhibent la synthèse des protéines en bloquant l'association des ARNt au ribosome lors de la traduction. Les tétracyclines d'origine naturelle produites par diverses souches de Streptomyces ont été découvertes pour la première fois dans les années 1940, et plusieurs tétracyclines semi-synthétiques, dont la doxycycline et la tigécycline, ont également été produites. Bien que les tétracyclines couvrent un large spectre d'agents pathogènes bactériens, les effets secondaires qui peuvent limiter leur utilisation incluent la phototoxicité, la décoloration permanente des dents en développement et la toxicité hépatique à fortes doses ou chez les patients atteints d'insuffisance rénale.
Inhibiteurs de synthèse des protéines qui lient la sous-unité 50S
Il existe plusieurs classes de médicaments antibactériens qui agissent en se liant à la sous-unité 50S des ribosomes bactériens. Les médicaments antibactériens macrolides ont une structure cyclique complexe et de grande taille et font partie d'une classe plus vaste de métabolites secondaires produits naturellement appelés polycétides, composés complexes produits par étapes par l'ajout répété d'unités à deux carbones par un mécanisme similaire à celui utilisé pour les graisses synthèse d'acides. Les macrolides sont des médicaments bactériostatiques à large spectre qui bloquent l'élongation des protéines en inhibant la formation de liaisons peptidiques entre des combinaisons spécifiques d'acides aminés. Le premier macrolide était l'érythromycine. Il a été isolé en 1952 à partir de Streptomyces erythreus et empêche la translocation. Les macrolides semi-synthétiques comprennent l'azithromycine et la télithromycine. Comparée à l'érythromycine, l'azithromycine a un spectre d'activité plus large, moins d'effets secondaires et une demi-vie significativement plus longue (1,5 heure pour l'érythromycine contre 68 heures pour l'azithromycine), ce qui permet une administration une fois par jour et un court traitement de 3 jours (formulation Zpac) pour la plupart des infections. La télithromycine est la première semi-synthétique de la classe des cétolides. Bien que la télithromycine présente une puissance et une activité accrues contre les agents pathogènes résistants aux macrolides, la Food and Drug Administration (FDA) des États-Unis a limité son utilisation au traitement de la pneumonie communautaire et exige l'étiquette « boîte noire » la plus forte pour le médicament en raison de sa grave hépatotoxicité.
Les lincosamides comprennent la lincomycine produite naturellement et la clindamycine semi-synthétique. Bien que structurellement distincts des macrolides, les lincosamides ont un mode d'action similaire à celui des macrolides en se liant à la sous-unité ribosomale 50S et en empêchant la formation de liaisons peptidiques. Les lincosamides sont particulièrement actifs contre les infections streptococciques et staphylococciques.
Le médicament chloramphénicol représente une autre classe d'antibactériens structurellement distincte qui se lient également au ribosome 50S, inhibant ainsi la formation de liaisons peptidiques. Le chloramphénicol, produit par Streptomyces venezuelae, a été découvert en 1947 ; en 1949, il est devenu le premier antibiotique à large spectre approuvé par la FDA. Bien qu'il s'agisse d'un antibiotique naturel, il est également facile à synthétiser et a été le premier médicament antibactérien produit synthétiquement en masse. En raison de sa production de masse, de sa couverture à large spectre et de sa capacité à pénétrer efficacement dans les tissus, le chloramphénicol a toujours été utilisé pour traiter un large éventail d'infections, de la méningite à la fièvre typhoïde en passant par la conjonctivite. Malheureusement, des effets secondaires graves, tels que le syndrome du bébé gris mortel et la suppression de la production de moelle osseuse, ont limité son rôle clinique. Le chloramphénicol provoque également une anémie de deux manières différentes. L'un des mécanismes consiste à cibler les ribosomes mitochondriaux au sein des cellules souches hématopoïétiques, provoquant une suppression réversible de la production de cellules sanguines dépendante de la dose. Une fois que le traitement de chloramphénicol est interrompu, la production de cellules sanguines revient à la normale. Ce mécanisme met en évidence la similitude entre les ribosomes 70S des bactéries et les ribosomes 70S au sein de nos mitochondries. Le deuxième mécanisme de l'anémie est idiosyncrasique (c'est-à-dire que le mécanisme n'est pas compris) et implique une perte létale irréversible de la production de cellules sanguines connue sous le nom d'anémie aplasique. Ce mécanisme d'anémie aplasique ne dépend pas de la dose et peut se développer après l'arrêt du traitement. En raison de problèmes de toxicité, l'utilisation du chloramphénicol chez l'homme est aujourd'hui rare aux États-Unis et se limite aux infections graves qui ne peuvent pas être traitées par des antibiotiques moins toxiques. Ses effets secondaires étant beaucoup moins graves chez les animaux, il est utilisé en médecine vétérinaire.
Les oxazolidinones, y compris le linézolide, constituent une nouvelle classe à large spectre d'inhibiteurs synthétiques de la synthèse des protéines qui se lient à la sous-unité ribosomale 50S des bactéries grampositives et gramnégatives. Cependant, leur mécanisme d'action semble quelque peu différent de celui des autres inhibiteurs de synthèse des protéines liant les sous-unités 50S déjà discutés. Ils semblent plutôt interférer avec la formation du complexe initiatique (association de la sous-unité 50S, de la sous-unité 30S et d'autres facteurs) pour la traduction, et ils empêchent la translocation de la protéine en croissance du site ribosomal A vers le site P. \(\PageIndex{3}\)Le tableau résume les inhibiteurs de synthèse des protéines.
| Cible moléculaire | Mécanisme d'action | Classe de médicaments | Médicaments spécifiques | Bactériostatique ou bactéricide | Spectre d'activité |
|---|---|---|---|---|---|
| Sous-unité 30S | Provoque des désappariements entre les codons et les anticodons, ce qui entraîne la formation de protéines défectueuses qui s'insèrent dans la membrane cytoplasmique | Aminoglycosides | Streptomycine, gentamicine, néomycine, kanamycine | Bactéricide | Large spectre |
| Bloque l'association des ARNt au ribosome | Tétracyclines | Tétracycline, doxycycline, tigécycline | Bactériostatique | Large spectre | |
| Sous-unité 50S | Bloque la formation de liaisons peptidiques entre les | Macrolides | Érythromycine, azithromycine, télithromycine | Bactériostatique | Large spectre |
| Lincosamides | Lincomycine, clindamycine | Bactériostatique | Spectre étroit | ||
| Non applicable | Chloramphénicol | Bactériostatique | Large spectre | ||
| Interfère avec la formation du complexe d'initiation entre les sous-unités 50S et 30S et d'autres facteurs. | Oxazolidinones | Linézolide | Bactériostatique | Large spectre |
Exercice\(\PageIndex{2}\)
Comparez et opposez les différents types d'inhibiteurs de la synthèse des protéines.
Inhibiteurs du fonctionnement des membranes
Un petit groupe d'antibactériens cible la membrane bactérienne comme mode d'action (Tableau\(\PageIndex{4}\)). Les polymyxines sont des antibiotiques polypeptidiques naturels découverts pour la première fois en 1947 en tant que produits de Bacillus polymyxa ; seules la polymyxine B et la polymyxine E (colistine) ont été utilisées en clinique. Ils sont lipophiles et possèdent des propriétés semblables à celles des détergents et interagissent avec le composant lipopolysaccharidique de la membrane externe des bactéries gram-négatives, perturbant ainsi leurs membranes externe et interne et tuant les cellules bactériennes. Malheureusement, le mécanisme de ciblage membranaire n'est pas une toxicité sélective, et ces médicaments ciblent et endommagent également la membrane des cellules du rein et du système nerveux lorsqu'ils sont administrés par voie systémique. En raison de ces effets secondaires graves et de leur faible absorption par le tube digestif, la polymyxine B est utilisée dans les pommades antibiotiques topiques en vente libre (par exemple, la néosporine), et la colistine orale était historiquement utilisée uniquement pour la décontamination intestinale afin de prévenir les infections causées par des microbes intestinaux patients immunodéprimés ou pour ceux qui subissent certaines chirurgies abdominales. Cependant, l'émergence et la propagation d'agents pathogènes multirésistants ont entraîné une augmentation de l'utilisation de la colistine par voie intraveineuse dans les hôpitaux, souvent comme médicament de dernier recours pour traiter des infections graves. La daptomycine, antibactérienne, est un lipopeptide cyclique produit par Streptomyces roseosporus qui semble agir comme les polymyxines, s'insérant dans la membrane cellulaire bactérienne et la perturbant. Cependant, contrairement à la polymyxine B et à la colistine, qui ciblent uniquement les bactéries gram-négatives, la daptomycine cible spécifiquement les bactéries gram-positives. Il est généralement administré par voie intraveineuse et semble bien toléré, présentant une toxicité réversible pour les muscles squelettiques.
| Mécanisme d'action | Classe de médicaments | Médicaments spécifiques | Spectre d'activité | Usage clinique |
|---|---|---|---|---|
| Interagit avec le lipopolysaccharide de la membrane externe des bactéries gram-négatives, tuant la cellule en perturbant éventuellement la membrane externe et la membrane cytoplasmique | Polymyxines | Polymyxine B | Spectre étroit contre les bactéries gram-négatives, y compris les souches multirésistantes | Préparations topiques pour prévenir les infections des plaies |
| Polymyxine E (colistine) | Spectre étroit contre les bactéries gram-négatives, y compris les souches multirésistantes | Dosage oral pour décontaminer les intestins afin de prévenir les infections chez les patients immunodéprimés ou les patients subissant une chirurgie/des interventions invasives. | ||
| Dosage par voie intraveineuse pour traiter les infections systémiques graves causées par des agents pathogènes multirésistants | ||||
| S'insère dans la membrane cytoplasmique des bactéries gram-positives, perturbant la membrane et tuant la cellule | Lipopeptide | Daptomycine | Spectre étroit contre les bactéries à Gram positif, y compris les souches multirésistantes | Infections compliquées de la peau et de la structure de la peau et bactériémie causées par des agents pathogènes à Gram positif, y compris le |
Exercice\(\PageIndex{3}\)
Comment les polymyxines inhibent-elles la fonction membranaire ?
Inhibiteurs de la synthèse des acides
Certains médicaments antibactériens agissent en inhibant la synthèse des acides nucléiques (Tableau\(\PageIndex{5}\)). Par exemple, le métronidazole est un membre semi-synthétique de la famille des nitroimidazoles qui est également un antiprotozoaire. Il interfère avec la réplication de l'ADN dans les cellules cibles. Le médicament rifampicine est un membre semi-synthétique de la famille des rifamycines et fonctionne en bloquant l'activité de l'ARN polymérase chez les bactéries. Les enzymes à ARN polymérase des bactéries sont structurellement différentes de celles des eucaryotes, ce qui entraîne une toxicité sélective contre les cellules bactériennes. Il est utilisé pour le traitement de diverses infections, mais son utilisation principale, souvent en cocktail avec d'autres médicaments antibactériens, est contre les mycobactéries responsables de la tuberculose. Malgré la sélectivité de son mécanisme, la rifampicine peut inciter les enzymes hépatiques à augmenter le métabolisme des autres médicaments administrés (antagonisme), ce qui entraîne une hépatotoxicité (toxicité hépatique) et une influence négative sur la biodisponibilité et l'effet thérapeutique des médicaments compagnons.
L'acide nalidixique est un membre de la famille des quinolones, un groupe d'antimicrobiens synthétiques. Il a été découvert en 1962 en tant que sous-produit lors de la synthèse de la chloroquine, un médicament antipaludique. L'acide nalidixique inhibe sélectivement l'activité de l'ADN gyrase bactérienne, bloquant ainsi la réplication de l'ADN. Les modifications chimiques apportées au squelette original des quinolones ont entraîné la production de fluoroquinolones, comme la ciprofloxacine et la lévofloxacine, qui inhibent également l'activité de l'ADN gyrase. La ciprofloxacine et la lévofloxacine sont efficaces contre un large éventail de bactéries gram-positives ou gram-négatives et font partie des antibiotiques les plus couramment prescrits pour traiter un large éventail d'infections, notamment les infections des voies urinaires, les infections respiratoires, les infections abdominales et les infections de la peau. Cependant, malgré leur toxicité sélective contre l'ADN gyrase, les effets secondaires associés aux différentes fluoroquinolones incluent la phototoxicité, la neurotoxicité, la cardiotoxicité, un dysfonctionnement du métabolisme du glucose et un risque accru de rupture des tendons.
| Mécanismes d'action | Classe de médicaments | Médicaments spécifiques | Spectre d'activité | Usage clinique |
|---|---|---|---|---|
| Inhibe l'activité de l'ARN polymérase bactérienne et bloque la transcription, tuant ainsi la cellule | Rifamycine | Rifampicine | Spectre étroit avec activité contre des bactéries gram-positives et un nombre limité de bactéries gram-négatives. Également actif contre Mycobacterium tuberculosis. | Thérapie combinée pour le traitement de la tuberculose |
| Inhibe l'activité de l'ADN gyrase et bloque la réplication de l'ADN, tuant ainsi la cellule | Fluoroquinolones | Ciprofloxacine, ofloxacine, moxifloxacine | Large spectre contre les bactéries gram-positives et gram-négatives | Grande variété d'infections cutanées et systémiques |
Exercice\(\PageIndex{4}\)
Pourquoi les inhibiteurs de la synthèse des acides nucléiques bactériens ne ciblent-ils pas les cellules hôtes ?
Inhibiteurs des voies métaboliques
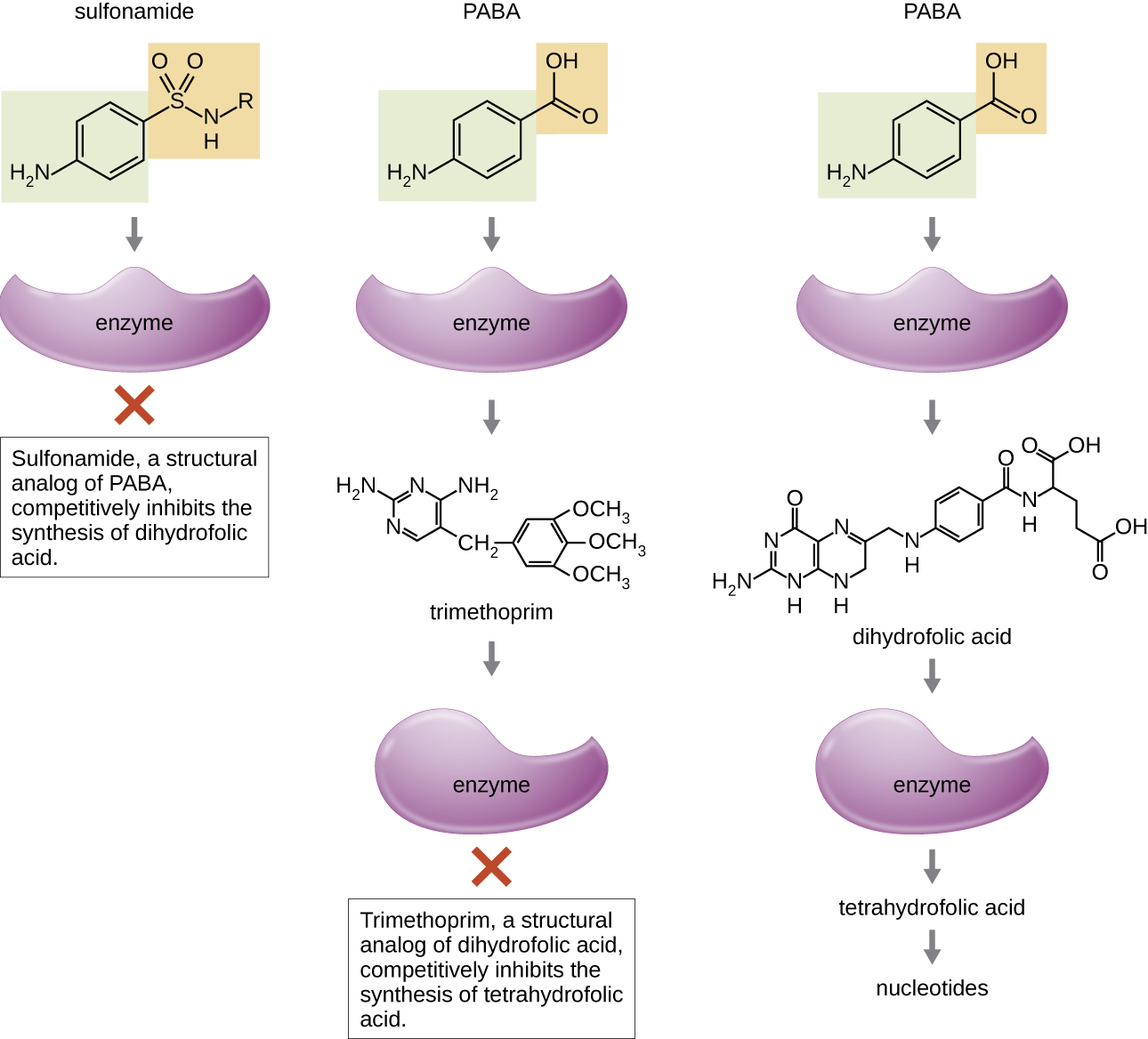
Certains médicaments synthétiques contrôlent les infections bactériennes en agissant comme des antimétabolites, des inhibiteurs compétitifs des enzymes métaboliques bactériennes (Tableau\(\PageIndex{6}\)). Les sulfamides (sulfamides) sont les plus anciens agents antibactériens synthétiques et sont des analogues structuraux de l'acide para -aminobenzoïque (PABA), un intermédiaire précoce de la synthèse de l'acide folique (Figure\(\PageIndex{4}\)). En inhibant l'enzyme impliquée dans la production de l'acide dihydrofolique, les sulfamides bloquent la biosynthèse bactérienne de l'acide folique et, par la suite, des pyrimidines et des purines nécessaires à la synthèse des acides nucléiques. Ce mécanisme d'action permet une inhibition bactériostatique de la croissance contre un large éventail d'agents pathogènes gram-positifs et gram-négatifs. Comme les humains obtiennent de l'acide folique à partir des aliments au lieu de le synthétiser par voie intracellulaire, les sulfamides sont sélectivement toxiques pour les bactéries. Cependant, les réactions allergiques aux sulfamides sont fréquentes. Les sulfones ont une structure similaire à celle des sulfamides mais ne sont pas couramment utilisés aujourd'hui, sauf pour le traitement de la maladie de Hansen (lèpre).
Le triméthoprime est un composé antimicrobien synthétique qui sert d'antimétabolite dans la même voie de synthèse de l'acide folique que les sulfamides. Cependant, le triméthoprime est un analogue structurel de l'acide dihydrofolique et inhibe une étape ultérieure de la voie métabolique (Figure\(\PageIndex{4}\)). Le triméthoprime est utilisé en association avec le sulfaméthoxazole, un sulfaméthoxazole, pour traiter les infections des voies urinaires, les otites et les bronchites. Comme indiqué, l'association du triméthoprime et du sulfaméthoxazole est un exemple de synergie antibactérienne. Lorsqu'il est utilisé seul, chaque antimétabolite ne fait que diminuer la production d'acide folique à un niveau tel qu'il se produit une inhibition bactériostatique de la croissance. Cependant, lorsqu'elle est utilisée en combinaison, l'inhibition des deux étapes de la voie métabolique réduit la synthèse de l'acide folique à un niveau létal pour la cellule bactérienne. En raison de l'importance de l'acide folique pendant le développement du fœtus, l'utilisation de sulfamides et de triméthoprime doit être soigneusement envisagée au début de la grossesse.
Le médicament isoniazide est un antimétabolite présentant une toxicité spécifique pour les mycobactéries et est utilisé depuis longtemps en association avec la rifampicine ou la streptomycine dans le traitement de la tuberculose. Il est administré sous forme de promédicament, nécessitant une activation par l'action d'une enzyme peroxydase bactérienne intracellulaire, formant de l'isoniazid-nicotinamide adénine dinucléotide (NAD) et de l'isoniazide-nicotinamide adénine dinucléotide phosphate (NADP), empêchant finalement la synthèse de l'acide mycolique, qui est essentiel pour les parois cellulaires des mycobactéries. Les effets secondaires possibles de l'utilisation de l'isoniazide incluent l'hépatotoxicité, la neurotoxicité et la toxicité hématologique (anémie).
| Voie métabolique cible | Mécanisme d'action | Classe de médicaments | Médicaments spécifiques | Spectre d'activité |
|---|---|---|---|---|
| Synthèse d'acide folique | Inhibe l'enzyme impliquée dans la production d'acide dihydrofolique | Sulfamides | Sulfaméthoxazole | Large spectre contre les bactéries gram-positives et gram-négatives |
| Sulfones | Dapsone | |||
| Inhibe l'enzyme impliquée dans la production d'acide tétrahydrofolique | Non applicable | Triméthoprime | Large spectre contre les bactéries gram-positives et gram-négatives | |
| Synthèse de l'acide mycolique | Interfère avec la synthèse de l'acide mycolique | Non applicable | Isoniazide | Spectre étroit contre Mycobacterium spp., y compris M. tuberculosis |
Exercice\(\PageIndex{5}\)
Comment les sulfamides et le triméthoprime ciblent-ils sélectivement les bactéries ?
Inhibiteur de l'ATP synthase
La bédaquiline, qui représente la classe de composés antibactériens synthétiques appelés diarylquinolones, utilise un nouveau mode d'action qui inhibe spécifiquement la croissance des mycobactéries. Bien que le mécanisme spécifique n'ait pas encore été élucidé, ce composé semble interférer avec la fonction des ATP synthases, peut-être en interférant avec l'utilisation du gradient d'ions hydrogène pour la synthèse de l'ATP par phosphorylation oxydative, ce qui entraîne une réduction de la production d'ATP. En raison de ses effets secondaires, notamment de l'hépatotoxicité et des arythmies cardiaques potentiellement mortelles, son utilisation est réservée aux cas graves de tuberculose qui ne seraient pas traités par ailleurs.
Orientation clinique : partie 2
En lisant attentivement les antécédents médicaux de Marisa, le médecin a remarqué que lors de son hospitalisation au Vietnam, elle avait été cathétérisée et avait reçu les médicaments antimicrobiens ceftazidime et métronidazole. Après avoir appris cela, le médecin a ordonné une tomodensitométrie de l'abdomen de Marisa pour exclure la possibilité d'une appendicite ; le médecin a également demandé des analyses de sang pour voir si elle avait un nombre élevé de globules blancs, et a ordonné une analyse d'urine et une culture d'urine pour rechercher la présence de globules blancs, de globules rouges et de bactéries .
L'échantillon d'urine de Marisa s'est révélé positif à la présence de bactéries, ce qui indique une infection des voies urinaires (IVU). Le médecin a prescrit de la ciprofloxacine. Entre-temps, son urine a été cultivée pour faire pousser la bactérie en vue d'autres tests.
Exercice\(\PageIndex{6}\)
- Quels types d'antimicrobiens sont généralement prescrits pour les infections urinaires ?
- Sur la base des médicaments antimicrobiens qui lui ont été administrés au Vietnam, lequel des antimicrobiens pour le traitement des infections urinaires serait inefficace selon vous ?
Concepts clés et résumé
- Les composés antibactériens présentent une toxicité sélective, principalement en raison des différences entre la structure des cellules procaryotes et eucaryotes.
- Les inhibiteurs de la synthèse de la paroi cellulaire, notamment les β-lactamines, les glycopeptides et la bacitracine, interfèrent avec la synthèse des peptidoglycanes, rendant les cellules bactériennes plus sujettes à la lyse osmotique.
- Il existe divers inhibiteurs de la synthèse des protéines bactériennes à large spectre qui ciblent sélectivement le ribosome procaryote 70S, y compris ceux qui se lient à la sous-unité 30S (aminoglycosides et tétracyclines) et d'autres qui se lient à la sous-unité 50S (macrolides, lincosamides, chloramphénicol et oxazolidinones).
- Les polymyxines sont des antibiotiques polypeptidiques lipophiles qui ciblent le composant lipopolysaccharidique des bactéries gram-négatives et perturbent finalement l'intégrité des membranes externe et interne de ces bactéries.
- Les inhibiteurs de synthèse des acides nucléiques, les rifamycines et les fluoroquinolones, ciblent respectivement la transcription de l'ARN bactérien et la réplication de l'ADN.
- Certains médicaments antibactériens sont des antimétabolites, agissant comme des inhibiteurs compétitifs des enzymes métaboliques bactériennes. Les sulfamides et le triméthoprime sont des antimétabolites qui interfèrent avec la synthèse bactérienne de l'acide folique. L'isoniazide est un antimétabolite qui interfère avec la synthèse de l'acide mycolique chez les mycobactéries.