
14.4 : Considérations cliniques
- Page ID
- 187922

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Objectifs d'apprentissage
- Expliquer les différences entre les modes d'action des médicaments qui ciblent les champignons, les protozoaires, les helminthes et les virus
Comme les champignons, les protozoaires et les helminthes sont eucaryotes, leurs cellules sont très similaires aux cellules humaines, ce qui rend plus difficile la mise au point de médicaments présentant une toxicité sélective. De plus, les virus se répliquent dans les cellules hôtes humaines, ce qui rend difficile la mise au point de médicaments sélectivement toxiques pour les virus ou les cellules infectées par des virus. Malgré ces défis, il existe des médicaments antimicrobiens qui ciblent les champignons, les protozoaires, les helminthes et les virus, et certains ciblent même plus d'un type de microbe. Le tableau\(\PageIndex{1}\), le tableau\(\PageIndex{2}\), le tableau\(\PageIndex{3}\) et le tableau\(\PageIndex{4}\) fournissent des exemples de médicaments antimicrobiens appartenant à ces différentes classes.
Médicaments antifongiques
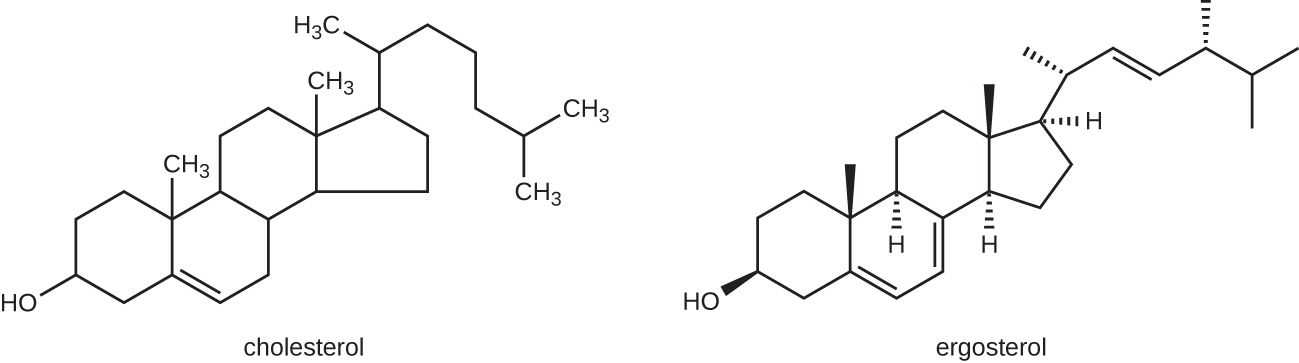
Le mode d'action le plus courant des médicaments antifongiques est la perturbation de la membrane cellulaire. Les antifongiques tirent parti des petites différences entre les champignons et les humains dans les voies biochimiques qui synthétisent les stérols. Les stérols jouent un rôle important dans le maintien d'une fluidité membranaire adéquate et, par conséquent, d'un bon fonctionnement de la membrane cellulaire. Pour la plupart des champignons, le stérol membranaire prédominant est l'ergostérol. Comme les membranes cellulaires humaines utilisent du cholestérol au lieu de l'ergostérol, les médicaments antifongiques qui ciblent la synthèse de l'ergostérol sont sélectivement toxiques (Figure\(\PageIndex{1}\)).
Les imidazoles sont des fongicides synthétiques qui perturbent la biosynthèse de l'ergostérol ; ils sont couramment utilisés dans des applications médicales et également en agriculture pour empêcher les semences et les cultures récoltées de se mouler. Parmi les exemples, citons le miconazole, le kétoconazole et le clotrimazole, qui sont utilisés pour traiter les infections cutanées fongiques telles que la teigne, en particulier le tinea pedis (pied d'athlète), le tinea cruris (démangeaisons du jock) et le tinea corporis. Ces infections sont généralement causées par des dermatophytes des genres Trichophyton, Epidermophyton et Microsporum. Le miconazole est également utilisé principalement pour le traitement des mycoses vaginales causées par le champignon Candida, et le kétoconazole est utilisé pour le traitement du teigne versicolor et des pellicules, qui peuvent tous deux être causés par le champignon Malassezia.
Les médicaments à base de triazole, y compris le fluconazole, inhibent également la biosynthèse de l'ergostérol. Cependant, ils peuvent être administrés par voie orale ou intraveineuse pour le traitement de plusieurs types d'infections systémiques à levures, notamment le muguet buccal et la méningite cryptococcique, qui sont toutes deux prévalentes chez les patients atteints du SIDA. Les triazoles présentent également une toxicité plus sélective que les imidazoles et sont associés à moins d'effets secondaires.
Les allylamines, une classe de médicaments antifongiques synthétiques structurellement différente, inhibent une étape plus précoce de la biosynthèse de l'ergostérol. L'allylamine la plus couramment utilisée est la terbinafine (commercialisée sous le nom de marque Lamisil), qui est utilisée localement pour le traitement des infections dermatophytiques de la peau telles que le pied d'athlète, la teigne et les démangeaisons du jock. Le traitement oral à la terbinafine est également utilisé pour le traitement de la mycose des ongles des mains et des pieds, mais il peut être associé au rare effet secondaire de l'hépatotoxicité.
Les polyènes sont une classe d'agents antifongiques produits naturellement par certaines bactéries actinomycètes du sol et sont structurellement apparentés aux macrolides. Ces grosses molécules lipophiles se lient à l'ergostérol dans les membranes cytoplasmiques des champignons, créant ainsi des pores. Les exemples courants incluent la nystatine et l'amphotéricine B. La nystatine est généralement utilisée comme traitement topique pour les mycoses de la peau, de la bouche et du vagin, mais peut également être utilisée pour les infections fongiques intestinales. Le médicament amphotéricine B est utilisé pour les infections fongiques systémiques telles que l'aspergillose, la méningite cryptococcique, l'histoplasmose, la blastomycose et la candidose. L'amphotéricine B était le seul antifongique disponible depuis plusieurs décennies, mais son utilisation est associée à certains effets secondaires graves, notamment une néphrotoxicité (toxicité rénale).
L'amphotéricine B est souvent utilisée en association avec la flucytosine, un analogue fluoré de la pyrimidine qui est converti par une enzyme spécifique aux champignons en un produit toxique qui interfère à la fois avec la réplication de l'ADN et la synthèse des protéines chez les champignons. La flucytosine est également associée à une hépatotoxicité (toxicité hépatique) et à une dépression médullaire.
Outre le ciblage de l'ergostérol dans les membranes des cellules fongiques, il existe quelques médicaments antifongiques qui ciblent d'autres structures fongiques (Figure\(\PageIndex{2}\)). Les échinocandines, y compris la caspofungine, sont un groupe de composés antifongiques produits naturellement qui bloquent la synthèse du β (1→3) glucane présent dans les parois cellulaires des champignons mais introuvable dans les cellules humaines. Cette classe de médicaments porte le surnom de « pénicilline pour les champignons ». La caspofungine est utilisée pour le traitement de l'aspergillose ainsi que des infections systémiques à levures.
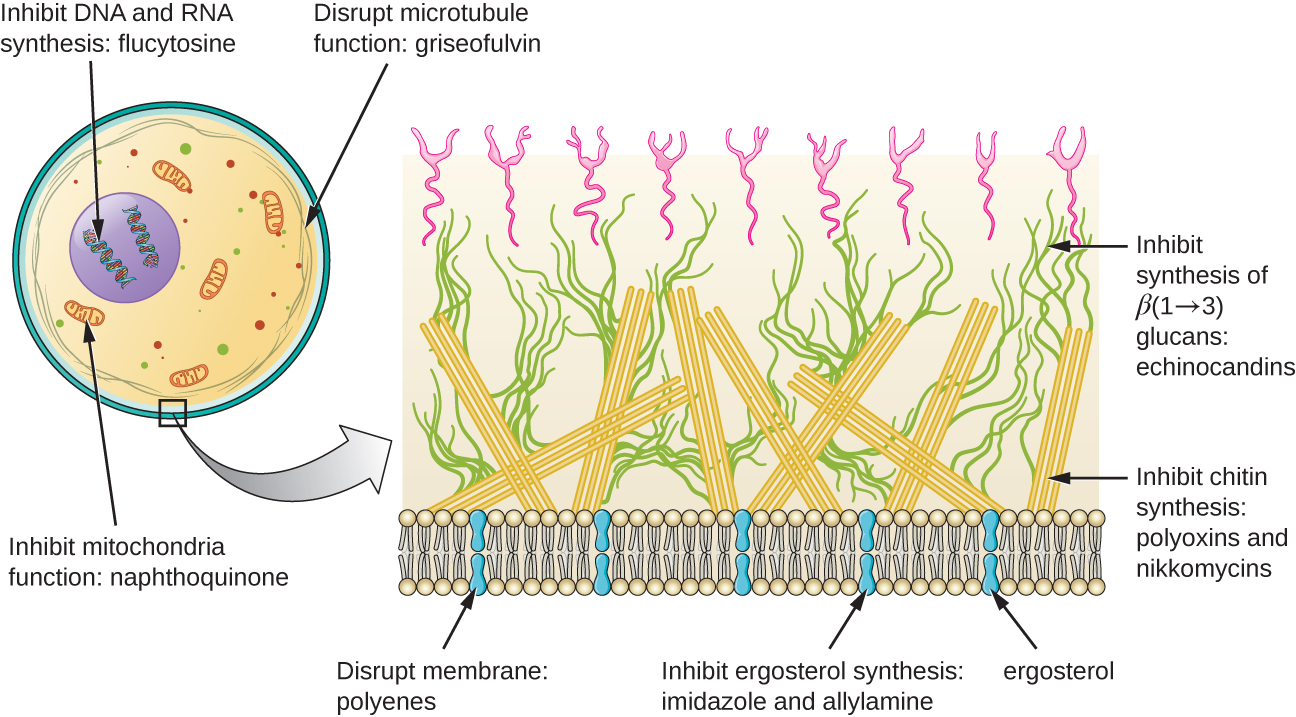
Bien que la chitine ne soit qu'un constituant mineur des parois cellulaires fongiques, elle est également absente des cellules humaines, ce qui en fait une cible sélective. Les polyoxines et les nikkomycines sont des antifongiques produits naturellement qui ciblent la synthèse de la chitine. Les polyoxines sont utilisées pour lutter contre les champignons à des fins agricoles, et la nikkomycine Z est actuellement en cours de développement pour être utilisée chez l'homme pour traiter les infections à levures et la fièvre de vallée (coccidioïdomycose), une maladie fongique répandue dans le sud-ouest des États-Unis. 1
On pense que la griséofulvine, un antifongique produit naturellement, perturbe spécifiquement la division cellulaire fongique en interférant avec les microtubules impliqués dans la formation des fuseaux lors de la mitose. C'était l'un des premiers antifongiques, mais son utilisation est associée à une hépatotoxicité. Il est généralement administré par voie orale pour traiter divers types d'infections dermatophytiques de la peau lorsque les autres traitements antifongiques topiques sont inefficaces.
Il existe quelques médicaments qui agissent comme antimétabolites contre les processus fongiques. Par exemple, l'atovaquone, un représentant de la classe des naphtoquinones, est un antimétabolite semi-synthétique des versions fongiques et protozoaires d'un cytochrome mitochondrial important pour le transport des électrons. Structurellement, c'est un analogue de la coenzyme Q, avec laquelle il entre en compétition pour la liaison électronique. Il est particulièrement utile pour le traitement de la pneumonie à pneumocystis causée par Pneumocystis jirovecii. L'association antibactérienne sulfaméthoxazole-triméthoprime agit également comme antimétabolite contre P. jirovecii.
Le tableau\(\PageIndex{1}\) montre les différentes classes thérapeutiques de médicaments antifongiques, classées par mode d'action, avec des exemples de chacune d'entre elles.
| Mécanisme d'action | Classe de médicaments | Médicaments spécifiques | Utilisations cliniques |
|---|---|---|---|
| Inhiber la synthèse d'ergostérol | Imidazoles | Miconazole, kétoconazole, clotrimazole | Infections cutanées fongiques et mycoses vaginales |
| Triazoles | Fluconazole | Mycoses systémiques, muguet buccal et méningite cryptococcique | |
| Allylamines | Terbinafine | Infections dermatophytiques de la peau (pied d'athlète, ver annulaire, démangeaisons) et infections des ongles des mains et des pieds | |
| Liez l'ergostérol dans la membrane cellulaire et créez des pores qui perturbent la membrane | Polyènes | Nystatine | Utilisé localement pour les infections à levures de la peau, de la bouche et du vagin ; également utilisé pour les infections fongiques de l'intestin |
| Amphotéricine B | Infections fongiques systémiques variées | ||
| Inhiber la synthèse des parois | Échinocandines | Caspofungine | Aspergillose et mycoses systémiques |
| Non applicable | Nikkomycine Z | Coccidioïdomycose (fièvre de la vallée) et infections à levures | |
| Inhiber les microtubules et la division cellulaire | Non applicable | Griséofulvine | Infections dermatophytiques de la peau |
Exercice\(\PageIndex{1}\)
En quoi la perturbation de la biosynthèse de l'ergostérol constitue-t-elle un mode d'action efficace pour les antifongiques ?
Traitement d'une infection fongique des poumons
Jack, un ingénieur de 48 ans, est séropositif mais est généralement en bonne santé grâce au traitement antirétroviral (TAR). Cependant, après une semaine de travail particulièrement intense, il a développé de la fièvre et une toux sèche. Il a supposé qu'il avait juste un rhume ou une légère grippe à cause d'un surmenage et n'y a pas beaucoup pensé. Cependant, après environ une semaine, il a commencé à ressentir de la fatigue, une perte de poids et un essoufflement. Il a décidé de consulter son médecin, qui a constaté que Jack avait un faible taux d'oxygénation du sang. Le médecin a demandé des analyses de sang, une radiographie pulmonaire et le prélèvement d'un échantillon de crachats provoqués à des fins d'analyse. Sa radiographie a révélé une nébulosité fine et plusieurs pneumatocèles (poches d'air à parois minces), indiquant une pneumonie à Pneumocystis (PCP), un type de pneumonie causé par le champignon Pneumocystis jirovecii. Le médecin de Jack l'a admis à l'hôpital et lui a prescrit du Bactrim, une combinaison de sulfaméthoxazole et de triméthoprime, à administrer par voie intraveineuse.
Le P. jirovecii est un champignon ressemblant à une levure dont le cycle de vie est similaire à celui des protozoaires. À ce titre, il a été classé comme protozoaire jusque dans les années 1980. Il vit uniquement dans les tissus pulmonaires des personnes infectées et se transmet d'une personne à l'autre, et de nombreuses personnes y sont exposées lorsqu'elles sont enfants. En général, P. jirovecii ne cause une pneumonie que chez les personnes immunodéprimées. Les personnes en bonne santé peuvent être porteuses du champignon dans leurs poumons sans aucun symptôme de maladie. La PCP est particulièrement problématique chez les patients séropositifs dont le système immunitaire est affaibli.
La PCP est habituellement traitée avec du Bactrim oral ou intraveineux, mais l'atovaquone ou la pentamidine (un autre médicament antiparasitaire) sont des alternatives. Si elle n'est pas traitée, la PCP peut progresser, entraînant un affaissement du poumon et une mortalité de près de 100 %. Même avec un traitement médicamenteux antimicrobien, la PCP est toujours responsable de 10 % des décès liés au VIH.
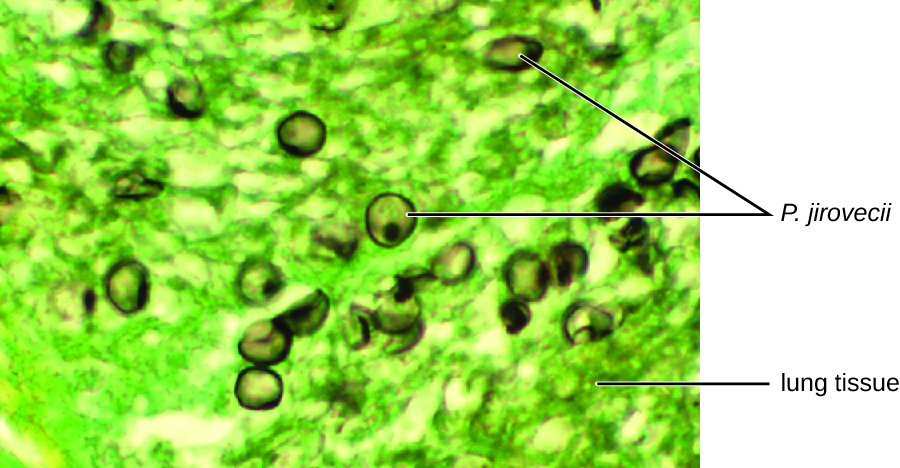
L'examen cytologique, à l'aide d'un test d'immunofluorescence directe (DFA), d'un frottis prélevé sur un échantillon de crachats de Jack a confirmé la présence de P. jirovecii (Figure\(\PageIndex{3}\)). De plus, les résultats des analyses sanguines de Jack ont révélé que son nombre de globules blancs avait chuté, ce qui le rendait plus vulnérable au champignon. Son médecin a revu son traitement antirétroviral et y a apporté des ajustements. Après quelques jours d'hospitalisation, Jack a été libéré pour poursuivre son traitement antimicrobien à domicile. Grâce aux ajustements apportés à sa thérapie antirétrovirale, le compte de CD4 de Jack a commencé à augmenter et il a pu retourner au travail.
Médicaments antiprotozoaires
Il existe quelques mécanismes par lesquels les médicaments antiprotozoaires ciblent les protozoaires infectieux (tableau\(\PageIndex{3}\)). Certains sont des antimétabolites, tels que l'atovaquone, le proguanil et les artémisinines. L'atovaquone, en plus d'être antifongique, bloque le transport d'électrons dans les protozoaires et est utilisée pour le traitement des infections à protozoaires, notamment le paludisme, la babésiose et la toxoplasmose. Le proguanil est un autre antimétabolite synthétique qui est transformé dans les cellules parasites en sa forme active, qui inhibe la synthèse de l'acide folique par les protozoaires. Il est souvent utilisé en association avec l'atovaquone, et l'association est commercialisée sous le nom de Malarone à la fois pour le traitement et la prévention du paludisme.
L'artémisinine, un antifongique d'origine végétale découvert pour la première fois par des scientifiques chinois dans les années 1970, est très efficace contre le paludisme. Les dérivés semi-synthétiques de l'artémisinine sont plus solubles dans l'eau que la version naturelle, ce qui les rend plus biodisponibles. Bien que le mécanisme d'action exact ne soit pas clair, les artémisinines semblent agir comme des promédicaments qui sont métabolisés par les cellules cibles pour produire des espèces réactives de l'oxygène (ROS) qui endommagent les cellules cibles. En raison de l'augmentation de la résistance aux médicaments antipaludiques, les artémisinines sont également couramment utilisées en association avec d'autres composés antipaludiques dans le cadre d'une polythérapie à base d'artémisinine (ACT).
Plusieurs antimétabolites sont utilisés pour le traitement de la toxoplasmose causée par le parasite Toxoplasma gondii. La sulfadiazine, un sulfamide synthétique, inhibe de manière compétitive une enzyme responsable de la production d'acide folique chez les parasites et peut être utilisée pour traiter le paludisme et la toxoplasmose. La pyriméthamine est un médicament synthétique qui inhibe une enzyme différente dans la voie de production d'acide folique et est souvent utilisée en association avec la sulfadoxine (un autre sulfamide) pour le traitement du paludisme ou en association avec la sulfadiazine pour le traitement de la toxoplasmose. Les effets secondaires de la pyriméthamine incluent une diminution de l'activité de la moelle osseuse pouvant entraîner une augmentation des ecchymoses et une baisse du nombre de globules rouges. Lorsque la toxicité est préoccupante, la spiramycine, un inhibiteur de la synthèse des protéines macrolides, est généralement administrée pour le traitement de la toxoplasmose.
Deux classes de médicaments antiprotozoaires interfèrent avec la synthèse des acides nucléiques : les nitroimidazoles et les quinoléines. Les nitroimidazoles, y compris le métronidazole semi-synthétique, dont il a déjà été question en tant que médicament antibactérien, et le tinidazole synthétique, sont utiles pour combattre une grande variété d'agents pathogènes protozoaires, tels que Giardia lamblia, Entamoeba histolytica et Trichomonas vaginalis . Lorsqu'ils sont introduits dans ces cellules dans des environnements pauvres en oxygène, les nitroimidazoles s'activent et cassent les brins d'ADN, interférant ainsi avec la réplication de l'ADN dans les cellules cibles. Malheureusement, le métronidazole est associé à la cancérogenèse (développement du cancer) chez l'homme.
La pentamidine est un autre type d'antiprotozoaire synthétique dont on a longtemps pensé qu'il interférait spécifiquement avec la réplication de l'ADN chez certains agents pathogènes. Il a toujours été utilisé pour le traitement de la maladie du sommeil (causée par le protozoaire Trypanosoma brucei) et de la leishmaniose (causée par des protozoaires du genre Leishmania) en Afrique, mais il s'agit également d'un traitement alternatif pour le champignon Pneumocystis. Certaines études indiquent qu'il se lie spécifiquement à l'ADN présent dans les kinétoplastes (kDNA ; longues structures semblables à des mitochondries propres aux trypanosomes), ce qui entraîne le clivage du kADN. Cependant, l'ADN nucléaire du parasite et de l'hôte n'est pas affecté. Il semble également se lier à l'ARNt, inhibant ainsi l'ajout d'acides aminés à l'ARNt, empêchant ainsi la synthèse des protéines. Les effets secondaires possibles de l'utilisation de pentamidine incluent un dysfonctionnement du pancréas et des lésions hépatiques.
Les quinoléines sont une classe de composés synthétiques apparentés à la quinine, qui est utilisée depuis longtemps contre le paludisme. On pense que les quinoléines interfèrent avec la détoxification de l'hème, qui est nécessaire à la décomposition efficace de l'hémoglobine par le parasite en acides aminés à l'intérieur des globules rouges. Les dérivés synthétiques chloroquine, quinacrine (également appelée mépacrine) et méfloquine sont couramment utilisés comme antipaludéens, et la chloroquine est également utilisée pour traiter l'amibiase généralement causée par Entamoeba histolytica. L'utilisation prophylactique à long terme de chloroquine ou de méfloquine peut provoquer des effets secondaires graves, notamment des hallucinations ou des problèmes cardiaques. Les patients présentant un déficit en glucose-6-phosphate déshydrogénase présentent une anémie sévère lorsqu'ils sont traités par la chloroquine.
| Mécanisme d'action | Classe de médicaments | Médicaments spécifiques | Utilisations cliniques |
|---|---|---|---|
| Inhiber le transport d'électrons dans les mitochondries | Naphtoquinone | Atovaquone | Paludisme, babésiose et toxoplasmose |
| Inhiber la synthèse de l'acide | Non applicable | Proquanil | Thérapie combinée avec l'atovaquone pour le traitement et la prévention du paludisme |
| Sulfamide | Sulfadiazine | Paludisme et toxoplasmose | |
| Non applicable | Pyriméthamine | Traitement combiné avec de la sulfadoxine (sulfamide) pour le traitement du paludisme | |
| Produit des espèces réactives nocives d'oxygène | Non applicable | Artémisinine | Thérapie combinée pour traiter le paludisme |
| Inhiber la synthèse | Nitroimidazoles | Métronidazole, tinidazole | Infections causées par Giardia lamblia, Entamoeba histolytica et Trichomonas vaginalis |
| Non applicable | Pentamidine | Maladie du sommeil et leishmaniose en Afrique | |
| Inhiber la désintoxication de l'hème | Quinoléines | Chloroquine | Paludisme et infections par E. histolytica |
| Mépacrine, méfloquine | Le paludisme |
Exercice\(\PageIndex{2}\)
Énumérez deux modes d'action des médicaments antiprotozoaires.
Médicaments antihelminthiques
Comme les helminthes sont des eucaryotes multicellulaires comme les humains, il est extrêmement difficile de mettre au point des médicaments présentant une toxicité sélective contre eux. Malgré cela, plusieurs classes efficaces ont été développées (Tableau\(\PageIndex{3}\)). Les benzimidazoles synthétiques, comme le mébendazole et l'albendazole, se lient à la β-tubuline helminthique, empêchant ainsi la formation de microtubules. Les microtubules présents dans les cellules intestinales des vers semblent particulièrement affectés, ce qui entraîne une réduction de l'absorption du glucose. Outre leur activité contre un large éventail d'helminthes, les benzimidazoles sont également actifs contre de nombreux protozoaires, champignons et virus, et leur utilisation pour inhiber la mitose et la progression du cycle cellulaire dans les cellules cancéreuses est à l'étude. 2 Les effets secondaires possibles de leur utilisation incluent des lésions hépatiques et une suppression de la moelle osseuse.
Les avermectines appartiennent à la famille des macrolides qui ont été découvertes pour la première fois à partir d'un isolat du sol japonais, Streptomyces avermectinius. Un dérivé semi-synthétique plus puissant de l'avermectine est l'ivermectine, qui se lie aux canaux chlorés dépendants du glutamate spécifiques aux invertébrés, y compris les helminthes, bloquant la transmission neuronale et provoquant la famine, la paralysie et la mort des vers. L'ivermectine est utilisée pour traiter les ascaris, notamment l'onchocercose (également appelée cécité des rivières, causée par le ver Onchocerca volvulus) et la strongyloïdose (causée par le ver Strongyloides stercoralis ou S. fuelleborni). L'ivermectine peut également traiter les insectes parasites tels que les acariens, les poux et les punaises de lit, et n'est pas toxique pour les humains.
Le niclosamide est un médicament synthétique utilisé depuis plus de 50 ans pour traiter les infections par le ténia. Bien que son mode d'action ne soit pas tout à fait clair, le niclosamide semble inhiber la formation d'ATP dans des conditions anaérobies et inhiber la phosphorylation oxydative dans les mitochondries de ses agents pathogènes cibles. Le niclosamide n'étant pas absorbé par le tractus gastro-intestinal, il peut atteindre des concentrations intestinales localisées élevées chez les patients. Récemment, il a été démontré qu'il avait également des activités antibactériennes, antivirales et antitumorales. 3 4 5
Un autre antihelminthique synthétique est le praziquantel, utilisé pour le traitement des ténias parasites et des douves du foie, et est particulièrement utile pour le traitement de la schistosomiase (causée par des douves sanguines de trois genres de Schistosoma). Son mode d'action demeure incertain, mais il semble provoquer un afflux de calcium dans le ver, provoquant des spasmes intenses et une paralysie du ver. Il est souvent utilisé comme alternative préférée au niclosamide dans le traitement des ténias lorsque l'inconfort gastro-intestinal limite l'utilisation du niclosamide.
Les thioxanthénones, une autre classe de médicaments synthétiques structurellement apparentés à la quinine, présentent une activité antischistosomique en inhibant la synthèse de l'ARN. La thioxanthénone, la lucanthone, et son métabolite, l'hycanthone, ont été les premiers à être utilisés en clinique, mais de graves effets secondaires neurologiques, gastro-intestinaux, cardiovasculaires et hépatiques ont conduit à leur arrêt. L'oxamniquine, un dérivé moins toxique de l'hycanthone, n'est efficace que contre S. mansoni, l'une des trois espèces connues pour provoquer la schistosomiase chez l'homme. Le praziquantel a été développé pour cibler les deux autres espèces de schistosomes, mais les préoccupations concernant l'augmentation de la résistance ont ravivé l'intérêt pour le développement de dérivés supplémentaires de l'oxamniquine pour cibler les trois espèces de schistosomes cliniquement importantes.
| Mécanisme d'action | Classe de médicaments | Médicaments spécifiques | Utilisations cliniques |
|---|---|---|---|
| Inhibe la formation de microtubules, réduisant ainsi l'absorption | Benzimidazoles | Mébendazole, albendazole | Variété d'helminthes |
| Bloquer la transmission neuronale, provoquant la paralysie et la famine | Avermectines | Ivermectine | Les ascaris, y compris la cécité des rivières et la strongyloïdose, et traitement des insectes parasites |
| Inhiber la production | Non applicable | Niclosamide | Infections intestinales par le ténia |
| Induquer un afflux | Non applicable | Praziquantel | Schistosomiase (douves sanguines) |
| Inhiber la synthèse | Thioxanthénones | Lucanthone, hycanthone, oxamniquine | Schistosomiase (douves sanguines) |
Exercice\(\PageIndex{3}\)
Pourquoi les antihelminthiques sont-ils difficiles à développer ?
Médicaments antiviraux
Contrairement à la structure complexe des champignons, des protozoaires et des helminthes, la structure virale est simple et se compose d'acide nucléique, d'une enveloppe protéique, d'enzymes virales et, parfois, d'une enveloppe lipidique. De plus, les virus sont des agents pathogènes intracellulaires obligatoires qui utilisent la machinerie cellulaire de l'hôte pour se répliquer. Ces caractéristiques rendent difficile la mise au point de médicaments présentant une toxicité sélective contre les virus.
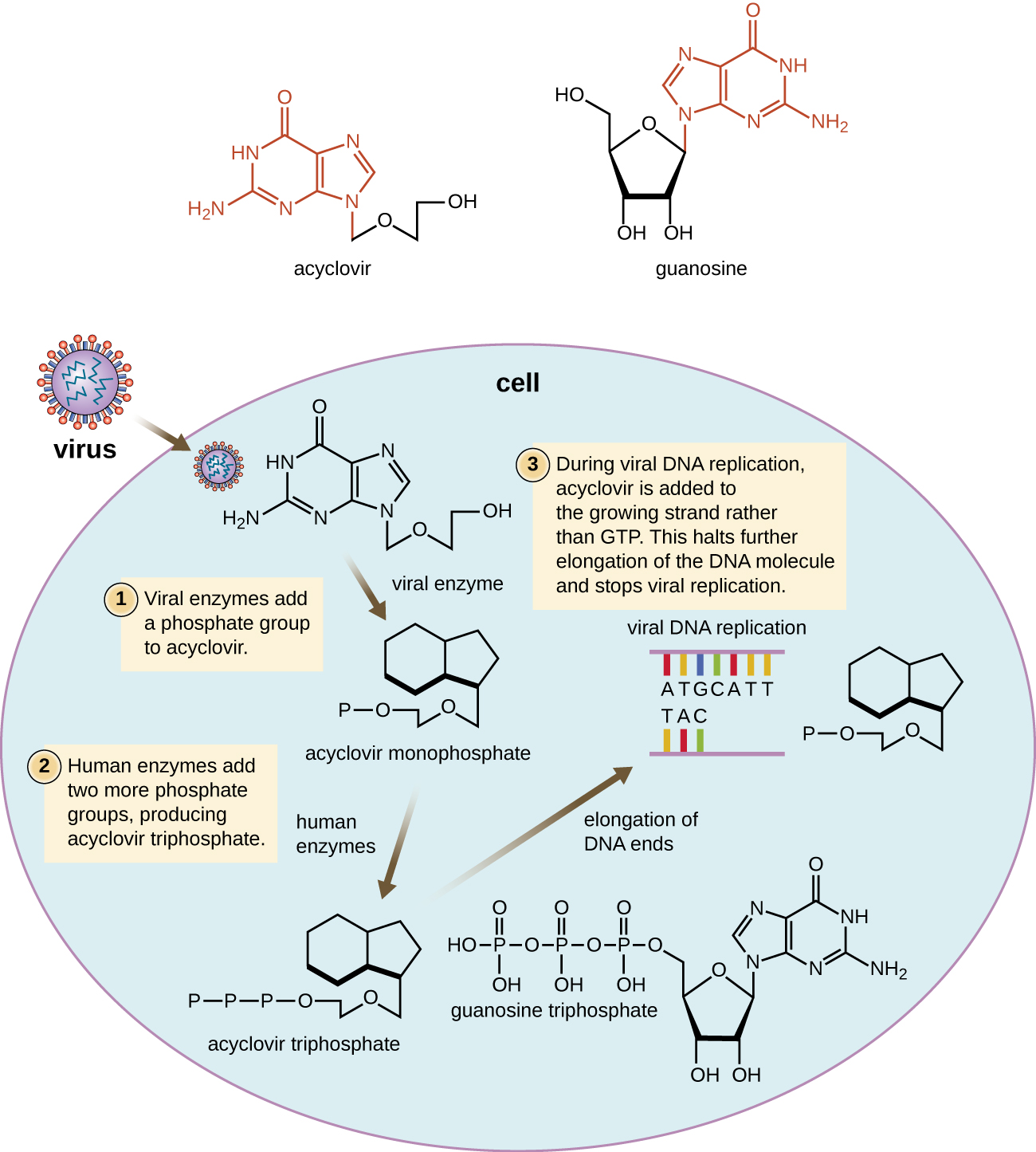
De nombreux médicaments antiviraux sont des analogues nucléosidiques et agissent en inhibant la biosynthèse des acides nucléiques. Par exemple, l'acyclovir (commercialisé sous le nom de Zovirax) est un analogue synthétique du nucléoside guanosine (Figure\(\PageIndex{4}\)). Elle est activée par l'enzyme virale de l'herpès simplex, la thymidine kinase et, lorsqu'elle est ajoutée à un brin d'ADN en croissance pendant la réplication, provoque la terminaison de la chaîne. Sa spécificité pour les cellules infectées par un virus provient à la fois de la nécessité d'une enzyme virale pour l'activer et de l'affinité accrue de la forme activée pour l'ADN polymérase virale par rapport à l'ADN polymérase de la cellule hôte. L'acyclovir et ses dérivés sont fréquemment utilisés pour le traitement des infections par le virus de l'herpès, notamment l'herpès génital, la varicelle, le zona, les infections par le virus d'Epstein-Barr et les infections par le cytomégalovirus. L'acyclovir peut être administré par voie topique ou systémique, selon l'infection. L'un des effets secondaires possibles de son utilisation comprend la néphrotoxicité. Le médicament adénine-arabinoside, commercialisé sous le nom de vidarabine, est un analogue synthétique de la désoxyadénosine dont le mécanisme d'action est similaire à celui de l'acyclovir. Il est également efficace pour le traitement de divers virus de l'herpès humain. Cependant, en raison des effets secondaires possibles liés à une baisse du nombre de globules blancs et à une neurotoxicité, le traitement par acyclovir est désormais préféré.
La ribavirine, un autre analogue synthétique de la guanosine, agit selon un mécanisme d'action qui n'est pas tout à fait clair. Il semble interférer à la fois avec la synthèse de l'ADN et de l'ARN, peut-être en réduisant les pools intracellulaires de guanosine triphosphate (GTP). La ribavarine semble également inhiber l'ARN polymérase du virus de l'hépatite C. Il est principalement utilisé pour le traitement des virus à ARN tels que l'hépatite C (en association avec l'interféron) et le virus respiratoire syncytial. Les effets secondaires possibles de l'utilisation de la ribavirine incluent l'anémie et des effets sur le développement des enfants à naître chez les patientes enceintes. Ces dernières années, un autre analogue nucléotidique, le sofosbuvir (Solvaldi), a également été développé pour le traitement de l'hépatite C. Le sofosbuvir est un analogue de l'uridine qui interfère avec l'activité de la polymérase virale. Il est fréquemment co-administré avec la ribavirine, avec et sans interféron.
L'inhibition de la synthèse des acides nucléiques n'est pas la seule cible des antiviraux synthétiques. Bien que le mode d'action de l'amantadine et de la rimantadine qui lui est apparentée ne soit pas tout à fait clair, ces médicaments semblent se lier à une protéine transmembranaire qui joue un rôle dans la fuite du virus de la grippe des endosomes. Le fait de bloquer la fuite du virus empêche également la libération de l'ARN viral dans les cellules hôtes et la réplication virale ultérieure. L'augmentation de la résistance a limité l'utilisation de l'amantadine et de la rimantadine dans le traitement de la grippe A. L'utilisation de l'amantadine peut entraîner des effets secondaires neurologiques, mais les effets secondaires de la rimantadine semblent moins graves. Il est intéressant de noter qu'en raison de leurs effets sur les substances chimiques du cerveau telles que la dopamine et le NMDA (N-méthyl D-aspartate), l'amantadine et la rimantadine sont également utilisées pour le traitement de la maladie de Parkinson.
Les inhibiteurs de la neuraminidase, notamment l'olsetamivir (Tamiflu), le zanamivir (Relenza) et le péramivir (Rapivab), ciblent spécifiquement les virus de la grippe en bloquant l'activité de la neuraminidase du virus grippal, empêchant ainsi la libération du virus par les cellules infectées. Ces trois antiviraux peuvent atténuer les symptômes de la grippe et raccourcir la durée de la maladie, mais leurs modes d'administration diffèrent : l'olsetamivir est administré par voie orale, le zanamivir est inhalé et le péramivir est administré par voie intraveineuse. La résistance à ces inhibiteurs de la neuraminidase semble encore minime.
Le pléconaril est un antiviral synthétique en cours de développement qui s'est révélé prometteur pour le traitement des picornavirus. L'utilisation du pléconaril pour le traitement du rhume banal causé par les rhinovirus n'a pas été approuvée par la FDA en 2002 en raison d'un manque d'efficacité prouvée, d'un manque de stabilité et d'une association avec des menstruations irrégulières. Son développement ultérieur à cette fin a été interrompu en 2007. Cependant, l'utilisation du pléconaril dans le traitement des complications potentiellement mortelles des entérovirus, telles que la méningite et la septicémie, est toujours à l'étude. Il est également étudié en vue de son utilisation dans l'éradication mondiale d'un entérovirus spécifique, la poliomyélite. 6 Le pléconaril semble agir en se liant à la capside virale et en empêchant la libération des particules virales à l'intérieur des cellules hôtes lors d'une infection virale.
Les virus dont le cycle de vie est complexe, comme le VIH, peuvent être plus difficiles à traiter. Tout d'abord, le VIH cible les globules blancs positifs aux CD4, qui sont nécessaires à une réponse immunitaire normale à l'infection. Ensuite, le VIH est un rétrovirus, c'est-à-dire qu'il convertit son génome d'ARN en une copie d'ADN qui s'intègre au génome de la cellule hôte, se cachant ainsi dans l'ADN de la cellule hôte. Troisièmement, la transcriptase inverse du VIH n'a aucune activité de relecture et introduit des mutations qui permettent le développement rapide d'une résistance aux antiviraux. Pour aider à prévenir l'apparition d'une résistance, une combinaison de médicaments antiviraux synthétiques spécifiques est généralement utilisée dans le traitement antirétroviral du VIH (Figure\(\PageIndex{5}\)).
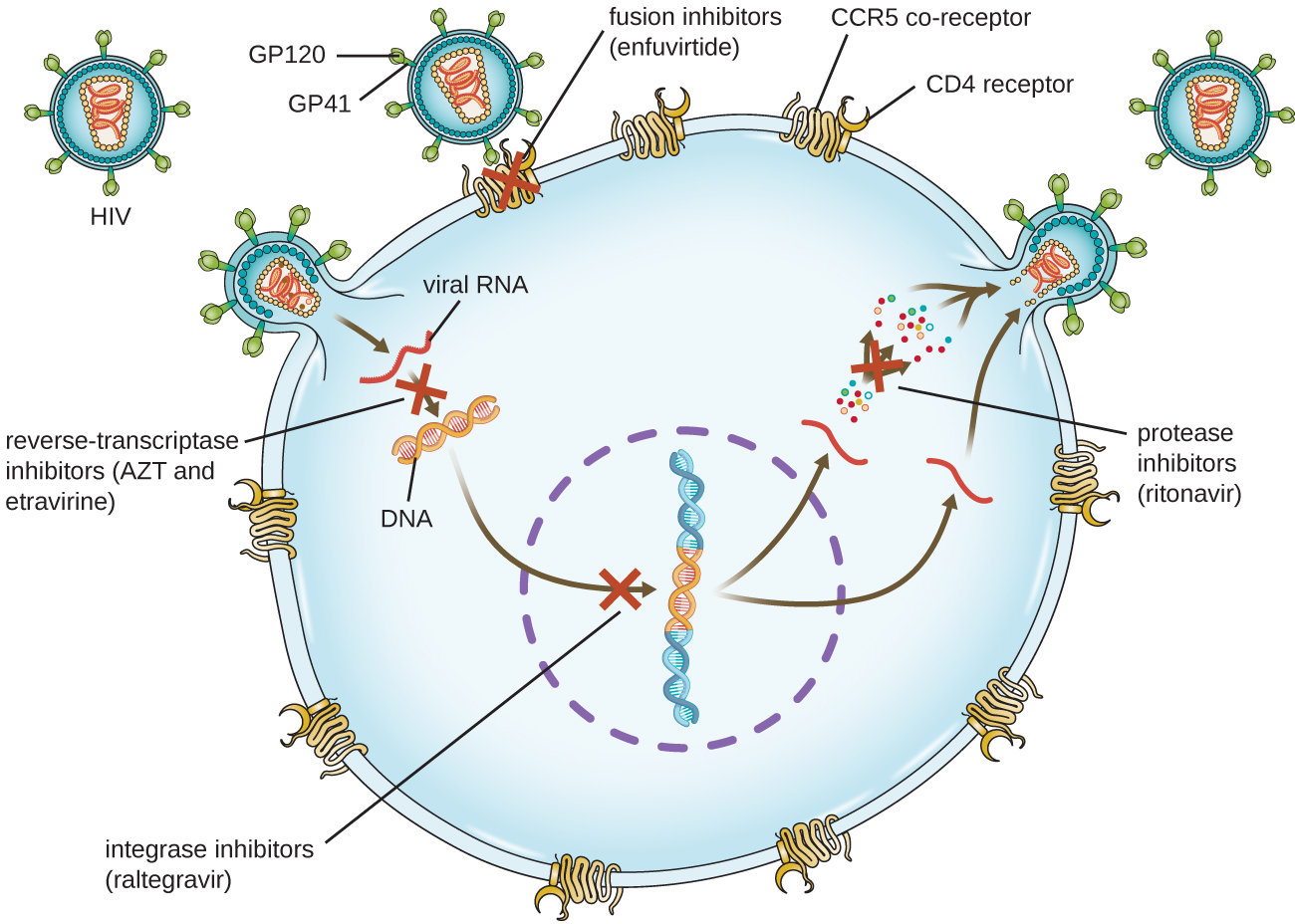
Les inhibiteurs de la transcriptase inverse bloquent l'étape précoce de la conversion du génome de l'ARN viral en ADN et peuvent inclure des inhibiteurs analogues nucléosidiques compétitifs (par exemple, azidothymidine/zidovudine ou AZT) et des inhibiteurs non compétitifs non nucléosidiques (par exemple, l'étravirine) qui se lient à la transcriptase inverse et provoquent une inactiver le changement de conformation. Les médicaments appelés inhibiteurs de protéase (par exemple, le ritonavir) bloquent le traitement des protéines virales et empêchent la maturation virale. Des inhibiteurs de protéase sont également en cours de développement pour le traitement d'autres types de virus. 7 Par exemple, le siméprévir (Olysio) a été approuvé pour le traitement de l'hépatite C et est administré en association avec la ribavirine et l'interféron. Les inhibiteurs de l'intégrase (par exemple, le raltégravir) bloquent l'activité de l'intégrase du VIH responsable de la recombinaison d'une copie d'ADN du génome viral dans le chromosome de la cellule hôte. Les autres classes de médicaments pour le traitement du VIH incluent les antagonistes du CCR5 et les inhibiteurs de fusion (par exemple, l'enfuviritide), qui empêchent la liaison du VIH au corécepteur de la cellule hôte (récepteur des chimiokines de type 5 [CCR5]) et la fusion de l'enveloppe virale avec la membrane de la cellule hôte, respectivement. Le tableau\(\PageIndex{4}\) montre les différentes classes thérapeutiques d'antiviraux, classées par mode d'action, avec des exemples de chacune d'entre elles.
| Mécanisme d'action | Médicament | Utilisations cliniques |
|---|---|---|
| Inhibition de la synthèse des acides nucléiques par des analogues nucléosi | Acyclovir | Infections herpétiques |
| Azidothymidine/zidovudine (AZT) | Infections au VIH | |
| Ribavirine | Infections par le virus de l'hépatite C et le virus respiratoire syncytial | |
| Vidarabine | Infections herpétiques | |
| Sofosbuvir | Infections au virus de l'hépatite | |
| Inhibition non compétitive non nucléosidique | Étravirine | Infections au VIH |
| Empêcher la fuite du virus par les endosomes | Amantadine, rimantadine | Infections au virus de la grippe |
| Inhiber la neuraminadase | Olétamivir, zanamivir, péramivir | Infections au virus de la grippe |
| Inhiber le décapage viral | Pléconaril | Infections graves à entérovirus |
| Inhibition de la protéase | Ritonavir | Infections au VIH |
| Siméprévir | Infections au virus de l'hépatite | |
| Inhibition de l'intégrase | Raltégravir | Infections au VIH |
| Inhibition de la fusion membranaire | Enfuviritide | Infections au VIH |
Exercice\(\PageIndex{4}\)
Pourquoi est-il difficile de traiter le VIH à l'aide d'antiviraux ?
Pour en savoir plus sur les différentes classes de médicaments antirétroviraux utilisés dans le traitement antirétroviral de l'infection par le VIH, explorez chacun des médicaments des classes de médicaments anti-VIH fournies par le ministère américain de la Santé et des Services sociaux sur ce site Web.
Concepts clés et résumé
- Comme les champignons, les protozoaires et les helminthes sont des organismes eucaryotes au même titre que les cellules humaines, il est plus difficile de développer des médicaments antimicrobiens qui les ciblent spécifiquement. De même, il est difficile de cibler les virus car les virus humains se répliquent à l'intérieur des cellules humaines.
- Les médicaments antifongiques interfèrent avec la synthèse de l'ergostérol, se lient à l'ergostérol pour perturber l'intégrité de la membrane cellulaire fongique ou ciblent des composants spécifiques de la paroi cellulaire ou d'autres protéines cellulaires.
- Les médicaments antiprotozoaires augmentent les niveaux cellulaires d'espèces réactives de l'oxygène, interfèrent avec la réplication de l'ADN protozoaire (ADN nucléaire contre ADN kADN, respectivement) et perturbent la détoxification de l'hème.
- Les antihelminthiques perturbent la formation des microtubules helminthiques et protozoaires ; bloquent les transmissions neuronales ; inhibent la formation anaérobie d'ATP et/ou la phosphorylation oxydative ; induisent un afflux de calcium dans les ténias, provoquant des spasmes et une paralysie ; et interfèrent avec la synthèse de l'ARN dans les schistosomes.
- Les médicaments antiviraux inhibent l'entrée du virus, inhibent le décapage viral, inhibent la biosynthèse des acides nucléiques, empêchent le virus de s'échapper des endosomes des cellules hôtes et empêchent la libération du virus par les cellules infectées.
- Comme il peut facilement muter pour devenir résistant aux médicaments, le VIH est généralement traité par une combinaison de plusieurs médicaments antirétroviraux, notamment des inhibiteurs de la transcriptase inverse, des inhibiteurs de protéase, des inhibiteurs de l'intégrase et des médicaments qui interfèrent avec la liaison et la fusion virales initier une infection.
Notes
- 1 Centres pour le contrôle et la prévention des maladies. « La fièvre de la vallée : la sensibilisation est essentielle ». www.cdc.gov/features/valleyfever/. Consulté le 1er juin 2016.
- 2 B. Chu et coll. « Un dérivé du benzimidazole présentant une activité antitumorale bloque l'activité de l'EGFR et de l'HER2 et régule à la hausse le DR5 dans les cellules cancéreuses du sein. » Mort cellulaire et maladie 6 (2015) :e1686
- 3 HEURES. Pan et coll. « Le niclosamide, un ancien antihelminthique, démontre une activité antitumorale en bloquant de multiples voies de signalisation des cellules souches cancéreuses. » Journal chinois du cancer 31 n° 4 (2012) :178—184.
- 4 F. Imperi et coll. « Une nouvelle vie pour un vieux médicament : le niclosamide, un anthelminthique, inhibe la détection du quorum de Pseudomonas aeruginosa. » Agents antimicrobiens et chimiothérapie 57 no 2 (2013) :996-1005.
- 5 A. Jurgeit et coll. « Le niclosamide est un vecteur de protons qui cible les endosomes acides avec de vastes effets antiviraux. » PLoS Pathogens 8 n° 10 (2012) :e1002976.
- 6 M. J. Abzug. « Les entérovirus : problèmes nécessitant des traitements. » Journal of Infection 68 no. S1 (2014) :108—14.
- 7 B. L. Pearlman. « Inhibiteurs de protéase pour le traitement de l'infection chronique par le génotype 1 de l'hépatite C : la nouvelle norme de soins. » Lancet Infectious Diseases 12 no. 9 (2012) :717—728.