
14.4:临床注意事项
- Page ID
- 200535

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)学习目标
- 解释靶向真菌、原生动物、蠕虫和病毒的药物作用模式之间的区别
由于真菌、原生动物和蠕虫是真核生物,因此它们的细胞与人体细胞非常相似,这使得开发具有选择性毒性的药物变得更加困难。 此外,病毒在人体宿主细胞内复制,这使得开发出对病毒或受病毒感染的细胞具有选择性毒性的药物变得困难。 尽管存在这些挑战,但仍有针对真菌、原生动物、蠕虫和病毒的抗微生物药物,有些甚至可以靶向不止一种微生物。 表\(\PageIndex{1}\)、表\(\PageIndex{2}\)\(\PageIndex{3}\)、表和表\(\PageIndex{4}\)提供了这些不同类别的抗微生物药物的示例。
抗真菌药物
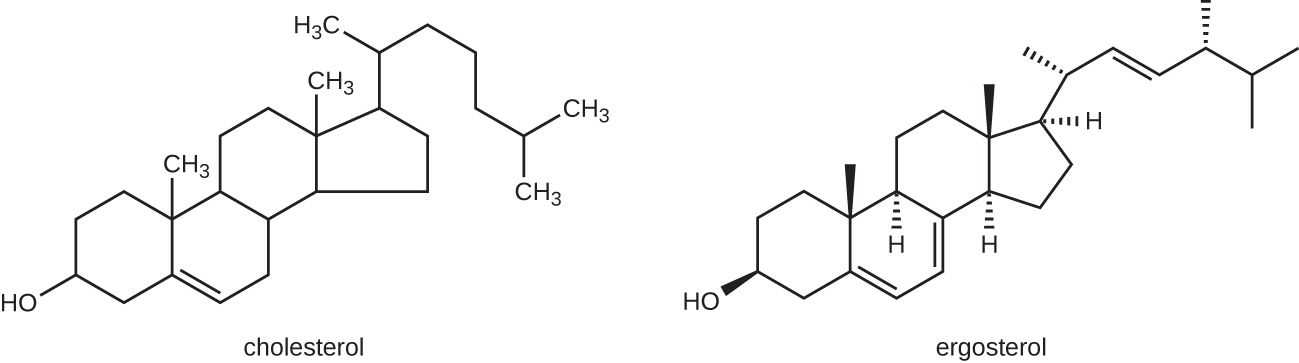
抗真菌药物最常见的作用方式是破坏细胞膜。 抗真菌药物利用真菌和人类在合成固醇的生化途径上的微小差异。 固醇对于维持适当的膜流动性以及细胞膜的正常功能非常重要。 对于大多数真菌来说,主要的膜固醇是麦角甾醇。 由于人体细胞膜使用胆固醇而不是麦角甾醇,因此靶向麦角甾醇合成的抗真菌药物具有选择性毒性(图\(\PageIndex{1}\))。
咪唑是破坏麦角甾醇生物合成的合成杀菌剂;它们通常用于医疗应用和农业,以防止种子和收获的农作物成型。 例子包括咪康唑、酮康唑和克霉唑,它们用于治疗皮肤真菌感染,例如癣,特别是足癣(脚癣)、tinea cruris(股痒)和 tinea corporis。 这些感染通常由 Trichophyt on、Epidermophyton 和 Mic rosp orum 属的皮肤真菌引起。 咪康唑还主要用于治疗由真菌念珠菌引起的阴道酵母菌感染,酮康唑用于治疗花斑癣和头皮屑,这两者都可能由真菌马拉色菌引起。
三唑药物,包括氟康唑,也抑制麦角甾醇的生物合成。 但是,它们可以口服或静脉注射,用于治疗几种类型的全身性酵母菌感染,包括口腔鹅口疮和隐球菌脑膜炎,这两者在艾滋病患者中都很普遍。 与咪唑相比,三唑类还表现出更高的选择性毒性,并且副作用也更少。
烯丙胺是一种结构不同的合成抗真菌药物,它抑制了麦角甾醇生物合成的早期步骤。 最常用的烯丙胺是特比萘芬(以Lamisil品牌销售),它局部用于治疗皮肤真菌性皮肤感染,例如脚癣、癣和股痒。 特比萘芬口服治疗也用于治疗指甲和脚趾甲真菌,但它可能与肝毒性的罕见副作用有关。
多烯是由某些放线菌土壤细菌自然产生的一类抗真菌药物,在结构上与大环内酯类有关。 这些大的亲脂分子与真菌细胞质膜中的麦角甾醇结合,从而形成毛孔。 常见的例子包括制霉菌素和两性霉素 B。制霉菌素通常用作皮肤、口腔和阴道酵母菌感染的局部治疗,但也可用于肠道真菌感染。 药物两性霉素B用于全身性真菌感染,如曲霉病、隐球菌脑膜炎、组织胞浆菌病、芽生菌病和念珠菌病。 Amphotericin B是几十年来唯一可用的抗真菌药物,但其使用会产生一些严重的副作用,包括肾毒性(肾脏毒性)。
Amphotericin B 通常与氟胞嘧啶联合使用,氟胞嘧啶是一种氟化嘧啶类似物,由真菌特异性酶转化为有毒产物,会干扰真菌中的 DNA 复制和蛋白质合成。 氟胞嘧啶还与肝毒性(肝毒性)和骨髓抑制有关。
除了靶向真菌细胞膜中的麦角甾醇外,还有一些针对其他真菌结构的抗真菌药物(图\(\PageIndex{2}\))。 echinocandins,包括 caspofungin,是一组天然产生的抗真菌化合物,可阻断存在于真菌细胞壁但不存在于人体细胞中的 β (1→3) 葡聚糖的合成。 该药物类别的昵称是 “用于真菌的青霉素”。 Caspofungin 用于治疗曲霉病和全身性酵母菌感染。
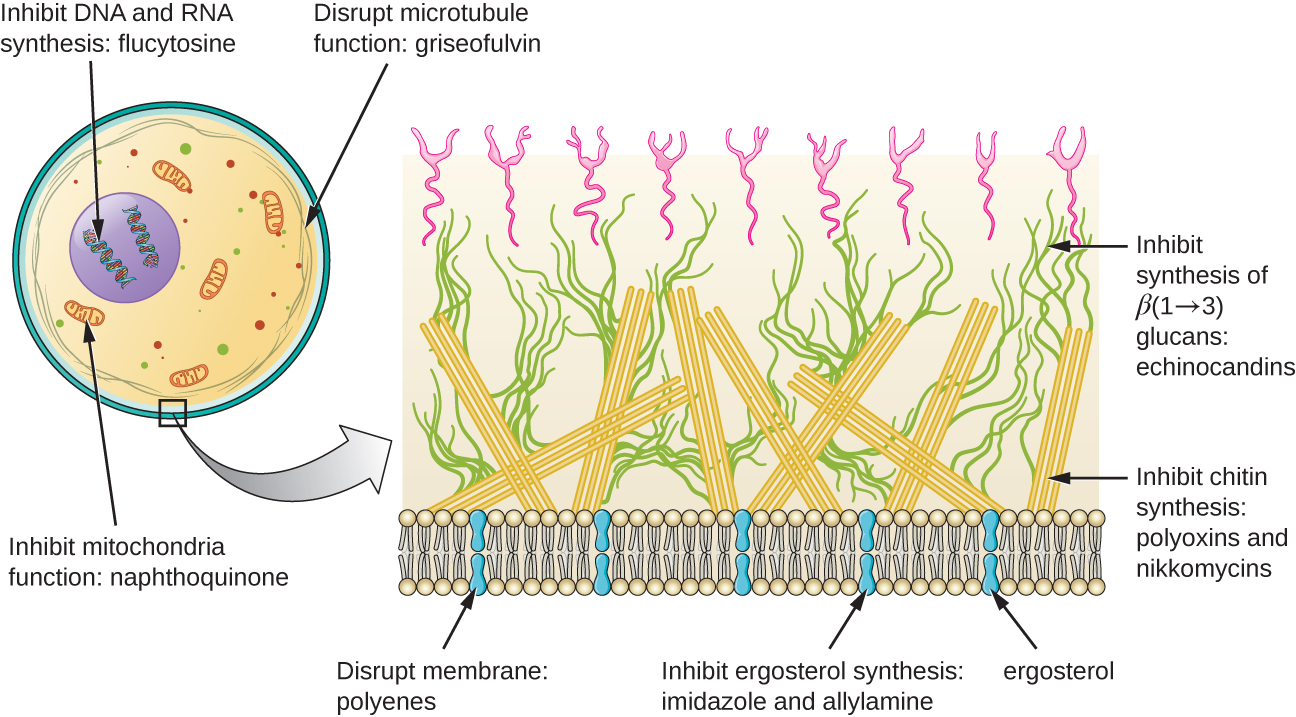
尽管甲壳素只是真菌细胞壁的次要成分,但它在人体细胞中也不存在,使其成为选择性靶标。 polyoxins 和 nikkomycins 是天然产生的抗真菌药物,靶向甲壳素合成。 Polyoxins 用于控制用于农业目的的真菌,而 nikkomycin Z 目前正在开发中,用于人类治疗酵母菌感染和谷热(球孢子菌病),这是一种在美国西南部流行的真菌病。 1
人们认为,天然产生的抗真菌灰黄霉素会干扰有丝分裂期间参与纺锤形成的微管,从而特别破坏真菌细胞的分裂。 它是最早的抗真菌药物之一,但其使用与肝毒性有关。 当其他局部抗真菌疗法无效时,它通常口服以治疗各种类型的皮肤真菌感染。
有一些药物可以作为抗真菌过程的抗代谢药。 例如,萘喹酮类药物的代表阿托伐酮是一种半合成抗代谢药,用于对电子传输很重要的线粒体细胞色素的真菌和原生动物版本。 从结构上讲,它是辅酶Q的类似物,它与辅酶Q竞争电子结合。 它对于治疗由吉罗韦氏肺孢子虫引起的肺孢子虫肺炎特别有用。 抗菌磺胺甲恶唑-甲氧苄啶组合也可以作为对抗吉罗韦氏杆菌的抗代谢药。
表中\(\PageIndex{1}\)显示了按作用方式分类的抗真菌药物的各种治疗类别,并附有每种药物的示例。
| 行动机制 | 药物类别 | 特定药物 | 临床用途 |
|---|---|---|---|
| 抑制麦角甾醇合成 | 咪唑类 | 咪康唑、酮康唑、克霉唑 | 皮肤真菌感染和阴道酵母菌感染 |
| 三唑类 | 氟康唑 | 全身性酵母菌感染、口腔鹅口疮和隐球菌脑膜炎 | |
| 烯丙胺 | 特比萘芬 | 皮肤真菌感染(脚臭、环虫、股痒)以及指甲和脚趾甲感染 | |
| 在细胞膜中结合麦角甾醇,形成破坏细胞膜的毛孔 | Polyenes | 制霉菌素 | 局部用于皮肤、口腔和阴道的酵母菌感染;也用于肠道真菌感染 |
| 两性霉素 B | 各种全身性真菌感染 | ||
| 抑制细胞壁合成 | Echinocandins | Caspofungin | 曲霉病和全身性酵母菌感染 |
| 不适用 | Nikkomycin Z | 球孢子菌病(谷热)和酵母菌感染 | |
| 抑制微管和细胞分裂 | 不适用 | Griseofulvin | 皮肤真菌性皮肤感染 |
练习\(\PageIndex{1}\)
破坏麦角甾醇的生物合成如何成为抗真菌药物的有效作用方式?
治疗肺部真菌感染
杰克是一名48岁的工程师,艾滋病毒呈阳性,但由于抗逆转录病毒疗法(ART),他总体健康。 但是,在特别紧张的一周工作之后,他出现了发烧和干咳。 他以为自己只是因为过度劳累而得了感冒或轻度流感,但没想太多。 但是,大约一周后,他开始感到疲劳、体重减轻和呼吸急促。 他决定去看医生,医生发现杰克的血氧含量很低。 医生下令进行血液检测、胸部X光检查,并收集诱导痰液样本进行分析。 他的 X 光片显示有细小的混浊和几个 pneumatoceles(薄壁空气袋),这表明肺孢子虫肺炎(PCP)是一种由真菌 Pneumocystis jirovecii 引起的肺炎。 杰克的医生将他送入医院,并开了由磺胺甲恶唑和甲氧苄啶混合的Bactrim处方进行静脉注射。
P. jirovecii 是一种酵母样真菌,其生命周期与原生动物相似。 因此,直到20世纪80年代,它才被归类为原生动物。 它仅存在于感染者的肺组织中,并在人与人之间传播,许多人在儿童时期就暴露在外。 通常,P. jirovecii 仅在免疫功能低下的人群中引起肺炎。 健康的人可能会将真菌带入肺部,而不会出现疾病症状。 在免疫系统受损的HIV患者中,五氯苯酚尤其成问题。
五氯苯酚通常通过口服或静脉注射 Bactrim 治疗,但阿托伐酮或喷他胺(另一种抗寄生虫药物)是替代品。 如果不加以治疗,五氯苯酚可能会进展,导致肺部塌陷和近 100% 的死亡率。 即使使用抗微生物药物治疗,五氯苯酚仍占艾滋病毒相关死亡的10%。
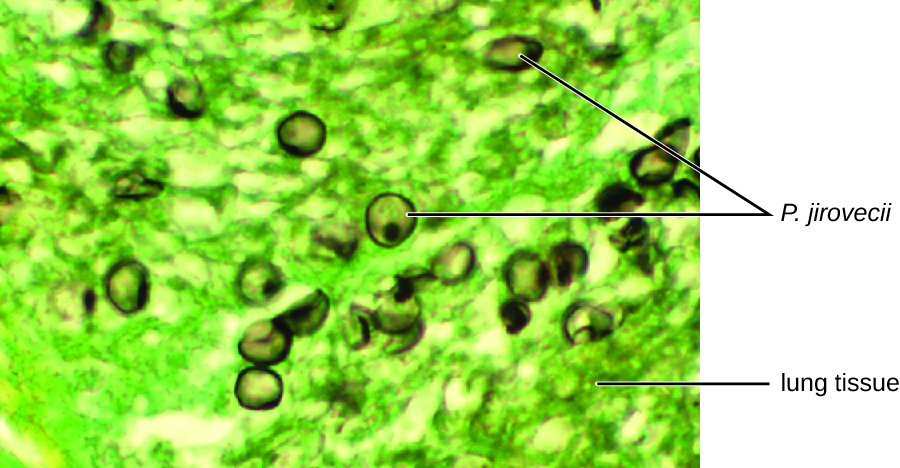
使用直接免疫荧光测定(DFA)对杰克痰液样本的涂片进行的细胞学检查证实了吉罗韦氏杆菌的存在(图\(\PageIndex{3}\))。 此外,杰克的血液检查结果显示他的白细胞数量下降了,使他更容易感染这种真菌。 他的医生审查了他的抗逆转录病毒疗法并进行了调整。 住院几天后,杰克获释继续在家接受抗微生物药物治疗。 随着抗逆转录病毒疗法的调整,杰克的CD4计数开始增加,他得以重返工作岗位。
抗原虫药物
抗原生动物药物靶向传染性原生动物有几种机制(表\(\PageIndex{3}\))。 有些是抗代谢药,例如阿托伐酮、丙胍和青蒿素。 Atovaquone 除了具有抗真菌作用外,还可以阻断原生动物的电子传输,并用于治疗原生动物感染,包括疟疾、巴贝斯虫病和弓形虫病。 Proguanil 是另一种合成抗代谢物,在寄生细胞中加工成活性形式,抑制原生动物叶酸的合成。 它通常与阿托伐酮联合使用,该组合物以马拉龙的名义销售,用于治疗和预防疟疾。
青蒿素是一种植物衍生的抗真菌药,由中国科学家在20世纪70年代首次发现,对疟疾非常有效。 青蒿素的半合成衍生物比天然版本更具水溶性,这使得它们更具生物利用度。 尽管确切的作用机制尚不清楚,但青蒿素似乎是前药,被靶细胞代谢,产生破坏靶细胞的活性氧(ROS)。 由于对抗疟药物的耐药性增加,青蒿素在青蒿素类复方疗法(ACT)中也常与其他抗疟化合物联合使用。
几种抗代谢药用于治疗由寄生虫弓形虫引起的弓形虫病。 合成磺胺药物磺胺二嗪竞争性地抑制寄生虫叶酸产生中的酶,可用于治疗疟疾和弓形虫病。 乙胺嘧啶是一种合成药物,可抑制叶酸产生途径中的另一种酶,通常与磺胺多辛(另一种磺胺药物)联合用于治疗疟疾或与磺胺二嗪联合用于治疗弓形虫病。 乙胺嘧啶的副作用包括骨髓活性降低,这可能导致瘀伤增加和红细胞计数降低。 当毒性成为问题时,通常使用大环内酯类蛋白质合成抑制剂螺旋霉素来治疗弓形虫病。
两类抗原生动物药物会干扰核酸的合成:硝基咪唑和喹啉。 硝基咪唑,包括先前作为抗菌药物讨论的半合成甲硝唑和合成替硝唑,可用于对抗各种原生动物病原体,例如贾第鞭毛虫、Entamoeba hi stolytica 和 Trich omonas vaginalis。 在低氧环境中引入这些细胞后,硝基咪唑会被激活并导致 DNA 链断裂,干扰靶细胞中的 DNA 复制。 不幸的是,甲硝唑与人类致癌(癌症的发展)有关。
另一种长期以来一直被认为会特别干扰某些病原体中DNA复制的合成抗原虫药物是喷他胺。 历史上,它曾用于治疗非洲昏睡病(由原生动物 Trypanosoma brucei 引起)和利什曼病(由利什曼原虫属的原生动物引起),但它也是真菌 Pneumocystis 的替代疗法。 一些研究表明,它与动质体(kDNA;锥虫独有的长线粒体样结构)中发现的DNA特异性结合,从而导致kDNA的分裂。 但是,寄生虫和宿主的核 DNA 仍未受到影响。 它似乎还与 tRNA 结合,抑制在 tRNA 中添加氨基酸,从而阻止蛋白质合成。 使用喷他胺可能产生的副作用包括胰腺功能障碍和肝损伤。
喹啉类是一类与奎宁相关的合成化合物,奎宁对疟疾的使用历史悠久。 喹啉被认为会干扰血红素排毒,而血红素排毒是寄生虫在红细胞内有效分解血红蛋白成氨基酸所必需的。 合成衍生物氯昆、喹那克林(也称为甲氟喹素)和甲氟喹通常用作抗疟药物,氯喹也用于治疗通常由组织溶解内阿米巴引起的阿米巴病。 长期预防性使用氯喹或甲氟喹可能会导致严重的副作用,包括幻觉或心脏问题。 葡萄糖-6-磷酸脱氢酶缺乏症患者在使用氯喹治疗时会出现严重的贫血。
| 行动机制 | 药物类别 | 特定药物 | 临床用途 |
|---|---|---|---|
| 抑制线粒体中的电子传输 | 萘喹酮 | Atovaquone | 疟疾、巴贝斯虫病和弓形虫病 |
| 抑制叶酸合成 | 不适用 | 丙喹尼 | 使用阿托伐酮联合疗法治疗和预防疟疾 |
| 磺酰胺 | 磺胺二嗪 | 疟疾和弓形虫病 | |
| 不适用 | 乙胺嘧啶 | 使用磺胺多辛(磺胺类药物)联合治疗疟疾 | |
| 产生有害的活性氧 | 不适用 | 青蒿素 | 治疗疟疾的联合疗法 |
| 抑制 DNA 合成 | 硝基咪唑 | 甲硝唑、替硝唑 | 由蓝氏贾第鞭毛虫、Entamoeba histolytica 和阴道滴虫引起的感染 |
| 不适用 | 喷他胺 | 非洲昏睡病和利什曼病 | |
| 抑制血红素排毒 | 喹啉类 | 氯 | 疟疾和溶组织大肠杆菌感染 |
| Mepacrine、甲氟喹等 | 疟疾 |
练习\(\PageIndex{2}\)
列出抗原虫药物的两种作用方式。
抗蠕虫药物
由于蠕虫像人类一样是多细胞真核生物,因此开发对它们具有选择性毒性的药物极具挑战性。 尽管如此,已经开发了几个有效的课程(表\(\PageIndex{3}\))。 合成苯并咪唑,如甲苯达唑和阿苯达唑,与蠕虫β-微管蛋白结合,防止微管形成。 蠕虫肠道细胞中的微管似乎特别受到影响,导致葡萄糖摄取减少。 除了对多种蠕虫的活性外,苯并咪唑还对许多原生动物、真菌和病毒具有活性,它们在抑制癌细胞中有丝分裂和细胞周期进展方面的用途正在研究中。 2 使用它们可能产生的副作用包括肝损伤和骨髓抑制。
阿维菌素是大环内酯家族的成员,最初是从日本土壤分离物 Streptomyces avermectinius 中发现的。 阿维菌素的一种更有效的半合成衍生物是伊维菌素,它与包括蠕虫在内的无脊椎动物特有的谷氨酸门控氯化物通道结合,阻断神经元传播,导致蠕虫饥饿、瘫痪和死亡。 伊维菌素用于治疗蚯蚓,包括盘尾丝虫病(也称为河盲症,由蠕虫 Onchocerc a volvulus 引起)和 stron gyloidiasis(由蠕虫 Strongyloides stercoralis 或 S. fuelleborni 引起)。 伊维菌素还可以治疗寄生昆虫,如螨虫、虱子和臭虫,对人类没有毒性。
Niclosamide 是一种合成药物,用于治疗绦虫感染已有 50 多年的历史。 尽管其作用模式尚不完全清楚,但氯硝柳胺似乎可以在厌氧条件下抑制ATP的形成,并抑制其靶病原体线粒体中的氧化磷酸化。 氯硝柳胺不会从胃肠道吸收,因此它可以在患者体内达到较高的局部肠道浓度。 最近,它已被证明还具有抗菌、抗病毒和抗肿瘤活性。 3 4 5
另一种合成抗蠕虫药物是吡喹酮,它用于治疗寄生绦虫和肝吸虫,对治疗血吸虫病(由血吸虫三属的血吸虫引起)特别有用。 它的作用方式尚不清楚,但它似乎会导致钙流入蠕虫中,导致蠕虫严重痉挛和瘫痪。 当胃肠道不适限制了氯硝柳胺的使用时,它通常被用作氯硝柳胺的首选替代品。
硫氧氰酮是另一类结构上与奎宁相关的合成药物,通过抑制RNA合成表现出抗血吸虫活性。 硫氧氰酮 lucanthone 及其代谢物 hycanthone 是最早在临床上使用的药物,但严重的神经、胃肠道、心血管和肝脏副作用导致其停药。 Oxamniquine 是一种毒性较低的 hycanth one 衍生物,仅对曼氏杆菌有效,曼氏杆菌是已知会导致人类血吸虫病的三种物种之一。 吡喹酮是为靶向其他两种血吸虫物种而开发的,但对耐药性增加的担忧重新激发了人们对开发其他奥沙米喹衍生物以靶向所有三种临床上重要的血吸虫物种的兴趣。
| 行动机制 | 药物类别 | 特定药物 | 临床用途 |
|---|---|---|---|
| 抑制微管形成,减少葡萄糖摄取 | 苯并咪唑 | 甲苯达唑、阿苯达唑 | 各种蠕虫感染 |
| 阻断神经元传播,导致瘫痪和饥饿 | 阿维菌素 | 伊维菌素 | 蝗虫病,包括河盲症和 strongyloidiasis,以及寄生昆虫的治疗 |
| 抑制 ATP 的产生 | 不适用 | Niclosamide | 肠道绦虫感染 |
| 诱导钙流入 | 不适用 | 吡喹酮 | 血吸虫病(血吸虫) |
| 抑制 RNA 合成 | 硫氧氰酮 | Lucanthone、hycanthone、oxamniquine | 血吸虫病(血吸虫) |
练习\(\PageIndex{3}\)
为什么抗蠕虫药物很难开发?
抗病毒药物
与真菌、原生动物和蠕虫的复杂结构不同,病毒结构很简单,由核酸、蛋白质外套、病毒酶,有时还包括脂质包膜组成。 此外,病毒是专用的细胞内病原体,利用宿主的细胞机制进行复制。 这些特性使得开发出对病毒具有选择性毒性的药物变得困难。
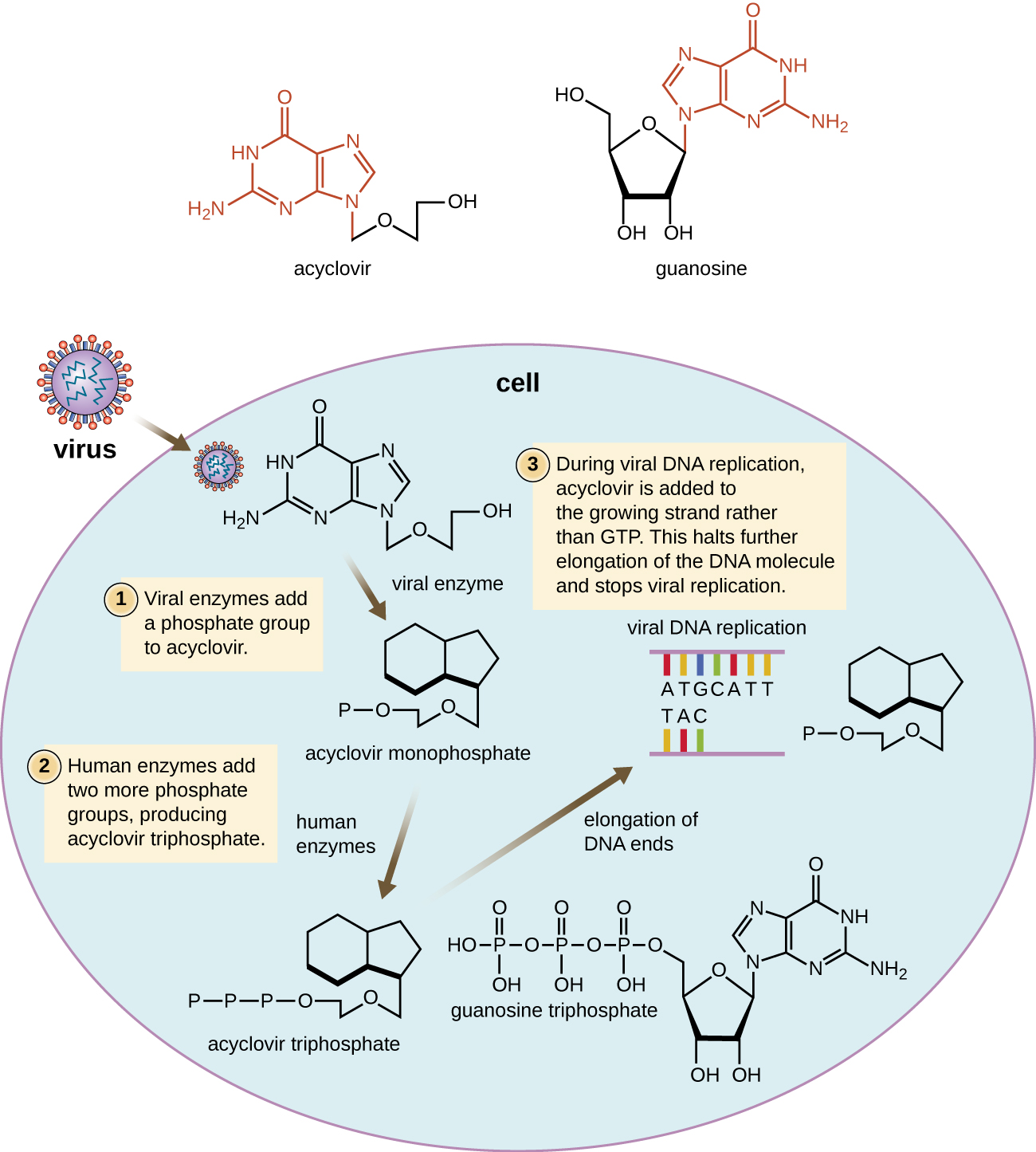
许多抗病毒药物都是核苷类似物,其作用是抑制核酸的生物合成。 例如,阿昔洛韦(以 Zovirax 的名义销售)是核苷鸟苷的合成类似物(图\(\PageIndex{4}\))。 它被单纯疱疹病毒酶胸苷激酶激活并在复制过程中添加到不断增长的DNA链中时,会导致链终止。 与宿主细胞 DNA 聚合酶相比,它对病毒感染细胞的特异性既来自于需要病毒酶来激活它,也源于活化形式对病毒 DNA 聚合酶的亲和力提高。 阿昔洛韦及其衍生物经常用于治疗疱疹病毒感染,包括生殖器疱疹、水痘、带状疱疹、爱泼斯坦-巴尔病毒感染和巨细胞病毒感染。 阿昔洛韦可以局部或全身给药,视感染情况而定。 其使用的一种可能的副作用包括肾毒性。 以维达拉滨的名义销售的腺嘌呤-阿拉伯糖苷是脱氧腺苷的合成类似物,其作用机制与阿昔洛韦相似。 它还可以有效治疗各种人类疱疹病毒。 但是,由于可能产生包括低白细胞计数和神经毒性的副作用,现在首选使用阿昔洛韦治疗。
利巴韦林是另一种合成鸟苷类似物,其作用机制尚不完全清楚。 它似乎会干扰DNA和RNA的合成,可能是通过减少细胞内三磷酸鸟苷(GTP)库来干扰。 利巴瓦林似乎还能抑制丙型肝炎病毒的RNA聚合酶。 它主要用于治疗RNA病毒,例如丙型肝炎(与干扰素联合治疗)和呼吸道合胞病毒。 使用利巴韦林可能产生的副作用包括贫血和对孕妇未出生婴儿的发育影响。 近年来,还开发了另一种核苷酸类似物索非布韦(Solvaldi),用于治疗丙型肝炎。索非布韦是一种干扰病毒聚合酶活性的尿苷类似物。 它通常与利巴韦林共同施用,有或没有干扰素。
抑制核酸合成不是合成抗病毒药物的唯一靶标。 尽管金刚烷胺及其相对金刚烷胺的作用模式尚不完全清楚,但这些药物似乎与一种跨膜蛋白结合,该蛋白参与了流感病毒从内窥体中逃脱。 阻断病毒逃逸还会阻止病毒 RNA 释放到宿主细胞中以及随后的病毒复制。 耐药性的增加限制了金刚烷胺和金刚他定治疗甲型流感的使用。使用金刚烷胺会导致神经系统副作用,但金刚烷定的副作用似乎不那么严重。 有趣的是,由于金刚烷胺和金刚烷胺对多巴胺和NMDA(N-甲基 D-天冬氨酸)等大脑化学物质的影响,金刚烷胺和金刚他定也用于治疗帕金森氏病。
神经氨酸酶抑制剂,包括奥司他韦(达菲)、扎那米韦(Relenza)和帕拉米韦(Rapivab),通过阻断流感病毒神经氨酸酶的活性,防止病毒从受感染细胞中释放,特异性靶向流感病毒。 这三种抗病毒药物可以减轻流感症状并缩短发病时间,但它们的给药方式不同:奥西他韦口服,扎那米韦吸入,帕拉米韦静脉注射。 对这些神经氨酸酶抑制剂的耐药性似乎仍然微乎其微。
Pleconaril是一种正在开发的合成抗病毒药物,显示出治疗小核糖核酸病毒的前景。 Pleconaril 用于治疗鼻病毒引起的普通感冒在 2002 年未获得 FDA 的批准,原因是缺乏经过验证的有效性、缺乏稳定性以及与月经不调有关。 它为此目的的进一步开发已于2007年停止。 但是,pleconaril 仍在研究是否用于治疗危及生命的肠道病毒并发症,例如脑膜炎和败血症。 还正在研究如何将其用于在全球消灭一种特定的肠道病毒脊髓灰质炎。 6 Pleconaril 的作用似乎是与病毒衣壳结合,防止病毒感染期间宿主细胞内的病毒颗粒脱皮。
生命周期复杂的病毒,例如艾滋病毒,可能更难治疗。 首先,HIV靶向CD4阳性白细胞,这是对感染产生正常免疫反应所必需的。 其次,HIV是一种逆转录病毒,这意味着它将其RNA基因组转化为整合到宿主细胞基因组中的DNA拷贝,从而隐藏在宿主细胞的DNA中。 第三,HIV逆转录酶缺乏校对活性,引入了导致抗病毒药物耐药性快速发展的突变。 为了帮助防止出现耐药性,在HIV的抗逆转录病毒疗法中通常使用特定的合成抗病毒药物的组合(图\(\PageIndex{5}\))。
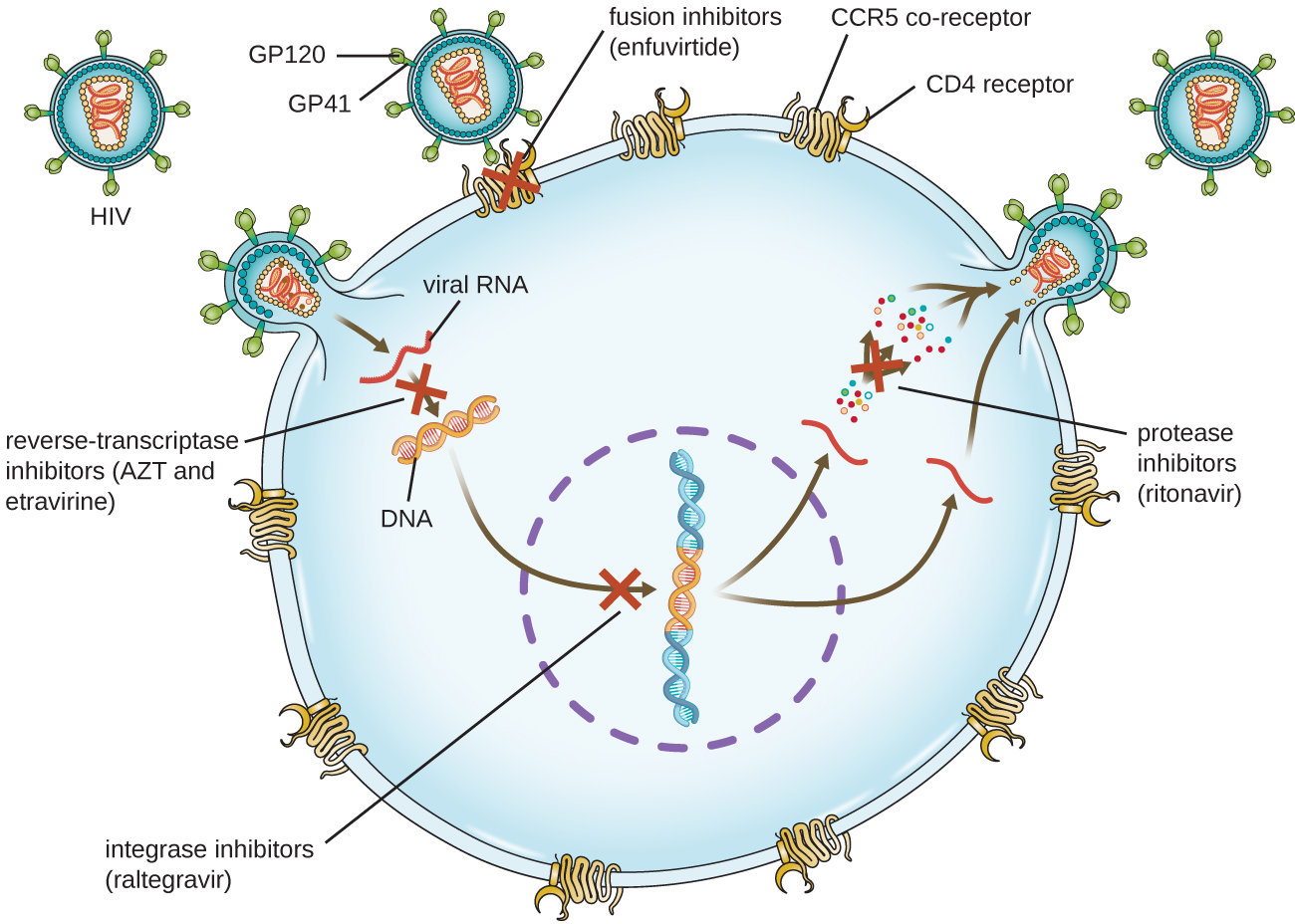
逆转录酶抑制剂阻断了将病毒 RNA 基因组转化为 DNA 的早期步骤,可以包括竞争性核苷类似物抑制剂(例如 azidothymidine/zidovudine 或 AZT)和非核苷非竞争性抑制剂(例如依曲韦林),它们结合逆转录酶并导致使构象变化失活。 称为蛋白酶抑制剂(例如利托那韦)的药物会阻断病毒蛋白的加工并阻止病毒成熟。 还正在开发用于治疗其他病毒类型的蛋白酶抑制剂。 7 例如,simeprevir(Olysio)已被批准用于治疗丙型肝炎,并在联合疗法中与利巴韦林和干扰素一起使用。 整合酶抑制剂(例如raltegravir)阻断负责将病毒基因组的DNA拷贝重组到宿主细胞染色体中的HIV整合酶的活性。 用于治疗HIV的其他药物类别包括CCR5拮抗剂和融合抑制剂(例如恩富维利肽),它们分别阻止HIV与宿主细胞共受体(5型趋化因子受体 [CCR5])结合以及病毒包膜与宿主细胞膜合并。 该表\(\PageIndex{4}\)显示了按作用方式分类的抗病毒药物的各种治疗类别,并附有每种药物的示例。
| 行动机制 | 毒品 | 临床用途 |
|---|---|---|
| 核苷类似物抑制核酸合成 | 阿昔洛韦 | 疱疹病毒感染 |
| Azidothymidine/Zidovudine (AZT) | 艾滋病毒感染 | |
| 利巴韦林 | 丙型肝炎病毒和呼吸道合胞病毒感染 | |
| Vidarabine | 疱疹病毒感染 | |
| 索氟布韦 | 丙型肝炎病毒感染 | |
| 非核苷非竞争性抑制 | 依曲韦林 | 艾滋病毒感染 |
| 抑制病毒从内窥体逃脱 | 金刚烷胺、金刚他定 | 流感病毒感染 |
| 抑制神经氨酸酶 | 奥司他韦、扎那米韦、帕拉米韦 | 流感病毒感染 |
| 抑制病毒脱皮 | Pleconaril | 严重的肠道病毒感染 |
| 抑制蛋白酶 | 利托那韦 | 艾滋病毒感染 |
| Simeprevir | 丙型肝炎病毒感染 | |
| 抑制整合酶 | Raltegravir | 艾滋病毒感染 |
| 抑制膜融合 | Enfuviritide | 艾滋病毒感染 |
练习\(\PageIndex{4}\)
为什么抗病毒药物很难治疗艾滋病毒?
要详细了解用于抗逆转录病毒治疗的艾滋病毒感染的各类抗逆转录病毒药物,请在本网站上浏览美国卫生与公共服务部提供的艾滋病毒药物类别中的每种药物。
关键概念和摘要
- 由于真菌、原生动物和蠕虫是像人体细胞一样的真核生物,因此开发专门针对它们的抗微生物药物更具挑战性。 同样,很难靶向病毒,因为人类病毒在人体细胞内部复制。
- 抗真菌药物会干扰麦角甾醇的合成,与麦角甾醇结合以破坏真菌细胞膜的完整性,或靶向细胞壁特异性成分或其他细胞蛋白。
- 抗原生动物药物会增加活性氧的细胞水平,干扰原生动物 DNA 复制(分别为核和 kDNA),并破坏血红素的排毒。
- 抗蠕虫药物破坏蠕虫和原生动物微管的形成;阻断神经元传播;抑制厌氧 ATP 的形成和/或氧化磷酸化;诱导钙流入绦虫,导致痉挛和瘫痪;干扰血吸虫中的 RNA 合成。
- 抗病毒药物抑制病毒进入,抑制病毒脱皮,抑制核酸生物合成,防止病毒从宿主细胞内窥体逃脱,并防止病毒从受感染细胞中释放。
- 由于HIV很容易突变而产生耐药性,因此通常使用多种抗逆转录药物联合治疗,其中可能包括逆转录酶抑制剂、蛋白酶抑制剂、整合酶抑制剂以及干扰病毒结合和融合的药物引发感染。
脚注
- 1 疾病控制与预防中心。 “Valley Fever:意识是关键。” www.cdc.gov/features/valleyfever/。 已于 2016 年 6 月 1 日访问。
- 2 B. Chu 等人。 “具有抗肿瘤活性的苯并咪唑衍生物可阻断乳腺癌细胞中的表皮生长因子和HER2活性,并上调DR5。” 细胞死亡与疾病 6 (2015): e1686
- 3 J.-X。 潘等 “氯硝柳胺是一种古老的抗蠕虫药物,它通过阻断癌症干细胞的多种信号通路表现出抗肿瘤活性。” 《中国癌症杂志》第 31 期第 4 期 (2012): 178—184。
- 4 F. Imperi 等人。 “旧药的新生活:驱虫药物 Niclosamide 抑制铜绿假单胞菌 Quorum Sens ing。” 抗微生物药物和化疗 57 no.2 (2013): 996-1005。
- 5 A. Jurgeit 等人。 “Niclosamide 是一种质子载体,靶向具有广泛抗病毒作用的酸性内窥体。” 《公共科学图书馆病原体》8 号第 10 号 (2012): e1002976。
- 6 M.J. Abzug。 “肠道病毒:需要治疗的问题。” 感染杂志 68 no. S1 (2014): 108—14。
- 7 B.L. Pearlman。 “用于治疗慢性丙型肝炎1型感染的蛋白酶抑制剂:新的护理标准。” 《柳叶刀传染病》第 12 期第 9 期 (2012): 717—728。