
14.3:靶向其他微生物的药物
- Page ID
- 200534

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)学习目标
- 描述与抑制细胞壁生物合成、蛋白质合成、膜功能、核酸合成和代谢途径的药物相关的作用机制。
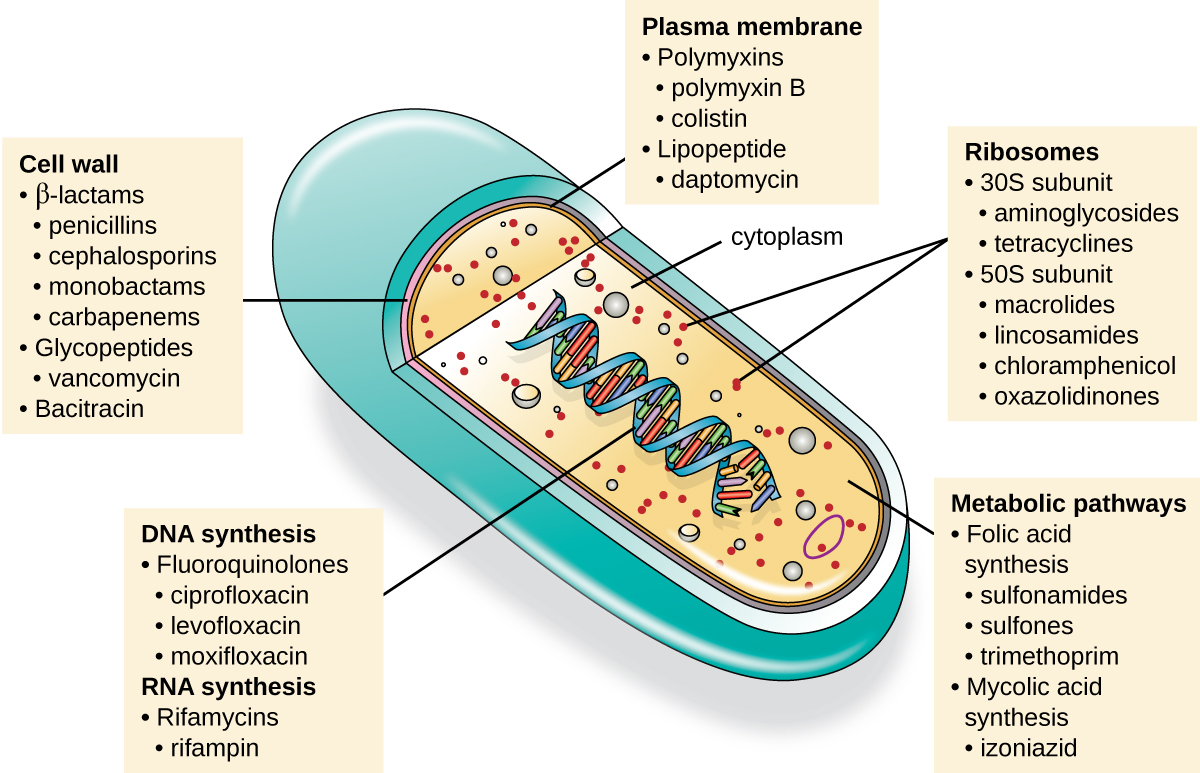
抗微生物药物的一个重要特性是选择性毒性,这意味着它有选择地杀死或抑制微生物靶标的生长,同时对宿主造成的伤害微乎其微或没有。 目前临床使用的大多数抗微生物药物都是抗菌药物,因为与真菌、寄生虫和病毒相比,原核细胞为选择性毒性提供了更多种类的独特靶标。 每类抗菌药物都有独特的作用模式(药物在细胞层面影响微生物的方式),这些在图\(\PageIndex{1}\)和表中进行了总结\(\PageIndex{1}\)。
| 行动模式 | 目标 | 药物类别 |
|---|---|---|
| 抑制细胞壁生物合成 | 青霉素结合蛋白 | β-内酰胺:青霉素、头孢菌素、单巴克胺、碳青霉烯类 |
| 肽聚糖亚单位 | 糖肽 | |
| 肽聚糖亚基转运 | 杆菌肽 | |
| 抑制蛋白质的生物合成 | 30S 核糖体亚单位 | 氨基糖苷类、四环素类 |
| 50S 核糖体亚单位 | 大环内酯类、lincosamides、氯霉素、oxazolidinones | |
| 破坏膜 | 脂多糖,内膜和外膜 | 多粘菌素 B、粘菌素、达托霉素 |
| 抑制核酸合成 | RNA | 利福霉素 |
| DNA | 氟喹诺酮类 | |
| 抗代谢药 | 叶酸合成酶 | 磺胺类药物、甲氧苄氨嘧啶 |
| 霉醇酸合成酶 | 异烟酸酰肼化物 | |
| 分枝杆菌三磷酸腺苷 (ATP) 合成酶抑制剂 | 分枝杆菌 ATP 合成酶 | 二芳基喹啉 |
细胞壁生物合成抑制剂
几种不同类别的抗菌药物会阻断肽聚糖的生物合成步骤,使细胞更容易受到渗透裂解的影响(表\(\PageIndex{2}\))。 因此,靶向细胞壁生物合成的抗菌药物的作用是杀菌的。 由于人体细胞不产生肽聚糖,因此这种作用模式是选择性毒性的一个很好的例子。
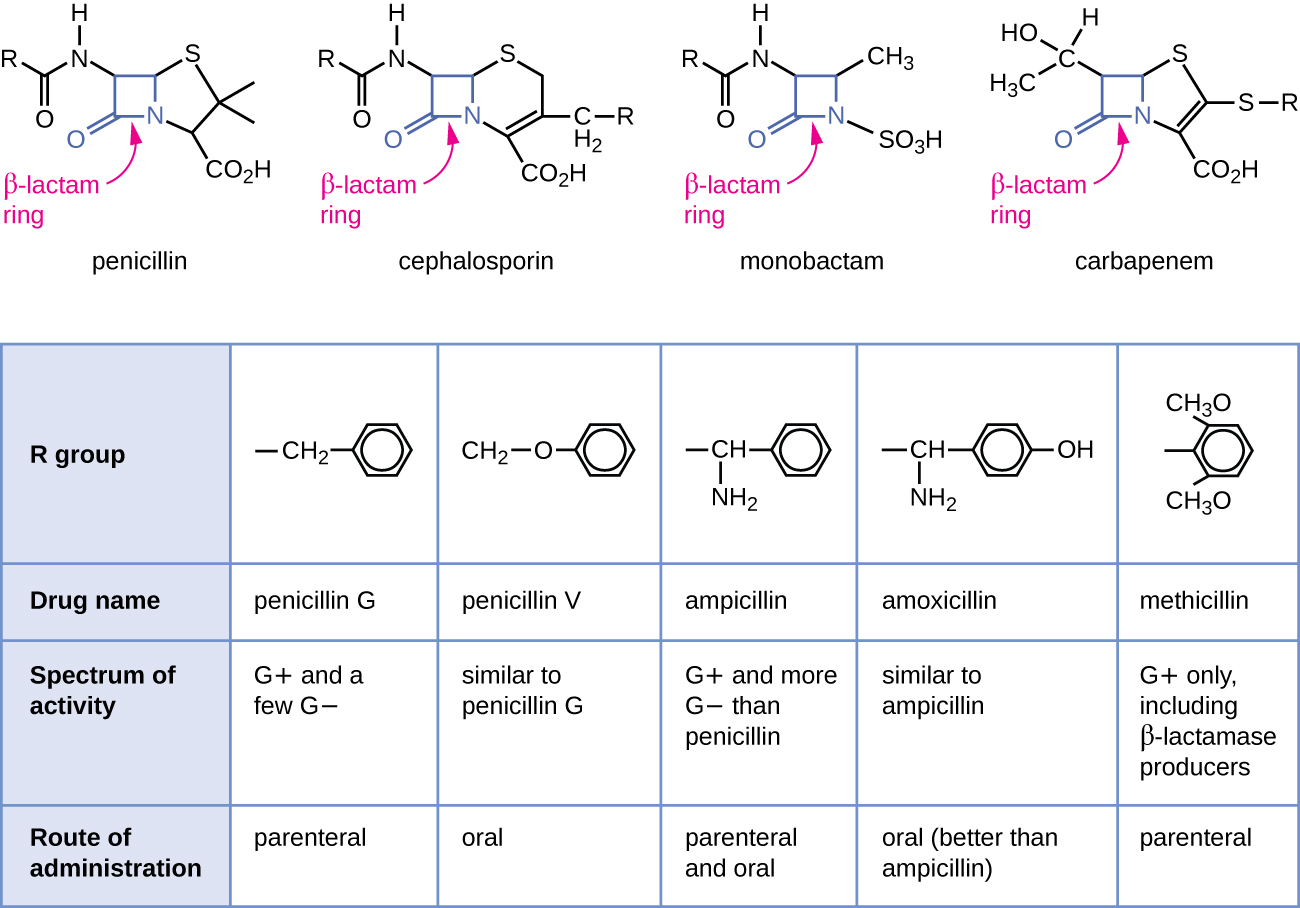
青霉素是第一种被发现的抗生素,是称为β-内酰胺类中的几种抗菌药物之一。 这组化合物包括青霉素、头孢菌素、单巴坦和碳青霉烯类,其特征是在药物分子的中心结构中存在β-内酰胺环(图\(\PageIndex{2}\))。 β-内酰胺抗菌剂在细菌细胞壁中生物合成新肽聚糖的过程中阻断肽链的交联。 他们之所以能够阻断这个过程,是因为β-内酰胺结构类似于肽聚糖亚单位成分的结构,后者被交联转肽酶(也称为青霉素结合蛋白(PBP))所识别。 尽管β-内酰胺环必须保持不变才能保持这些药物的抗菌活性,但R组的战略化学变化使开发出各种各样的半合成β-内酰胺药物,这些药物的效力更高,活性范围扩大,半衰期更长剂量等特征。
青霉素 G 和青霉素 V 是来自真菌的天然抗生素,主要对革兰氏阳性细菌病原体和一些革兰氏阴性细菌病原体(例如多杀性巴氏杆菌)具有活性。 图中\(\PageIndex{2}\)总结了一些青霉素的半合成发育情况。 在青霉素 G 中添加氨基团(-NH 2)会产生氨基青霉素(即氨苄青霉素和阿莫西林),它们对更多革兰氏阴性病原体的活性范围有所增加。 此外,在阿莫西林中添加羟基 (-OH) 可提高酸稳定性,从而改善口服吸收。 甲氧西林是一种半合成的青霉素,旨在解决使其他青霉素失活的酶(青霉素酶)的传播。 将青霉素 G 的 R 组改为更大体积的二甲氧基苯基可以保护 β-内酰胺环免受青霉素酶的酶破坏,从而为我们提供了第一种抗青霉素酶的青霉素。
与青霉素类似,头孢菌素含有 β-内酰胺环(图\(\PageIndex{2}\)),可阻断青霉素结合蛋白的转肽酶活性。 但是,头孢菌素的β-内酰胺环与六元环融合在一起,而不是青霉素中的五元环。 这种化学差异使头孢菌素对β-内酰胺酶的酶失活具有更高的抵抗力。 药物头孢菌素C最初是在20世纪50年代从真菌C ephalosporium acremonium 中分离出来的,其对革兰氏阳性细菌的活性范围与青霉素相似,但对革兰氏阴性细菌的活性比青霉素多。 另一个重要的结构差异是,头孢菌素 C 具有两个 R 基团,而青霉素只有一个 R 组,这为半合成头孢菌素的化学变化和发育提供了更大的多样性。 半合成头孢菌素家族比青霉素大得多,这些药物主要根据其活性谱分为几代人,从第一代窄谱头孢菌素到广谱第四代头孢菌素。 已经开发出一种新的第五代头孢菌素,它对耐甲氧西林金黄色葡萄球菌(MRSA)具有活性。
碳青霉烯类和单体胺还具有β-内酰胺环作为其核心结构的一部分,它们抑制青霉素结合蛋白的转肽酶活性。 临床上唯一使用的单巴坦是阿曲南。 它是一种窄谱抗菌剂,仅对革兰氏阴性细菌具有活性。 相比之下,碳青霉烯家族包括各种半合成药物(亚胺培南、美罗培南和多利培南),它们对革兰氏阳性和革兰氏阴性细菌病原体具有非常广谱的活性。
万古霉素是一类称为糖肽的化合物的成员,是在20世纪50年代发现的,是一种来自放线菌 Amycolatopsis oriental is的天然抗生素。 与β-内酰胺类似,万古霉素抑制细胞壁生物合成并具有杀菌作用。 但是,与β-内酰胺相比,万古霉素的结构与细胞壁肽聚糖亚基的结构不相似,也不会直接使青霉素结合蛋白失活。 相反,万古霉素是一种非常大、复杂的分子,它与细胞壁前体的肽链末端结合,形成结构阻塞,阻止细胞壁亚基掺入肽聚糖不断增长的N-乙酰氨基葡萄糖和N-乙酰穆拉米酸(NAM-NAG)骨干中结构(转糖基化)。 万古霉素还能在结构上阻断转肽。 万古霉素对革兰氏阳性细菌病原体具有杀菌作用,但由于无法穿透保护性外膜,它对革兰氏阴性细菌没有活性。
药物杆菌肽由一组结构相似的肽类抗生素组成,最初从枯草芽孢杆菌中分离出来。 杆菌肽阻断特定细胞膜分子的活性,该分子负责肽聚糖前体从细胞质向细胞外部的移动,最终阻止它们融入细胞壁。 杆菌肽对多种细菌有效,包括皮肤上发现的革兰氏阳性生物,例如葡萄球菌和链球菌。 尽管在某些情况下可以口服或肌肉注射,但杆菌肽已被证明具有肾毒性(对肾脏有害)。 因此,在局部药膏(例如Neosporin)中,它更常与新霉素和多粘菌素联合使用。
| 行动机制 | 药物类别 | 特定药物 | 天然或半合成 | 活动范围 |
|---|---|---|---|---|
| 直接与 PBP 相互作用并抑制 transpeptidase 活性 | 青霉素 | 青霉素 G、青霉素 V | 自然 | 针对革兰氏阳性和少数革兰氏阴性细菌的窄光谱 |
| 氨苄西林、阿莫西林 | 半合成的 | 针对革兰氏阳性细菌的窄光谱但革兰氏阴性谱增加 | ||
| 甲氧西林 | 半合成的 | 仅针对革兰氏阳性细菌,包括产生青霉素酶的菌株 | ||
| 头孢菌素 | 头孢菌素 C | 自然 | 窄谱与青霉素类似,但革兰氏阴性谱增加 | |
| 第一代头孢菌素 | 半合成的 | 与头孢菌素 C 相似的窄光谱 | ||
| 第二代头孢菌素 | 半合成的 | 与第一代相比,光谱较窄,但革兰氏阴性谱增加 | ||
| 第三代和第四代头孢菌素 | 半合成的 | 广谱抗革兰氏阳性和革兰氏阴性细菌,包括一些 β-内酰胺酶产生者 | ||
| 第五代头孢菌素 | 半合成的 | 广谱抗革兰氏阳性和革兰氏阴性细菌,包括金黄色葡萄球菌 | ||
| Monobactams | Aztreonam | 半合成的 | 针对革兰氏阴性细菌(包括一些 β-内酰胺酶生产者)的窄谱检测 | |
| 碳青霉烯类 | 亚胺培南、美罗培南、多利培南 | 半合成的 | 对抗革兰氏阳性和革兰氏阴性细菌的 β-内酰胺谱最广,包括许多 β-内酰胺酶产生者 | |
| 与 peptidoglycan 亚基的肽链结合,阻断转糖基化和转肽的大分子 | 糖肽 | 万古霉素 | 自然 | 仅针对革兰氏阳性细菌(包括耐多药菌株)的窄光谱 |
| 阻断肽聚糖亚基在细胞质膜上的转运 | 杆菌肽 | 杆菌肽 | 自然 | 广谱抗革兰氏阳性和革兰氏阴性细菌 |
练习\(\PageIndex{1}\)
描述β-内酰胺的作用方式。
蛋白质生物合成抑制剂
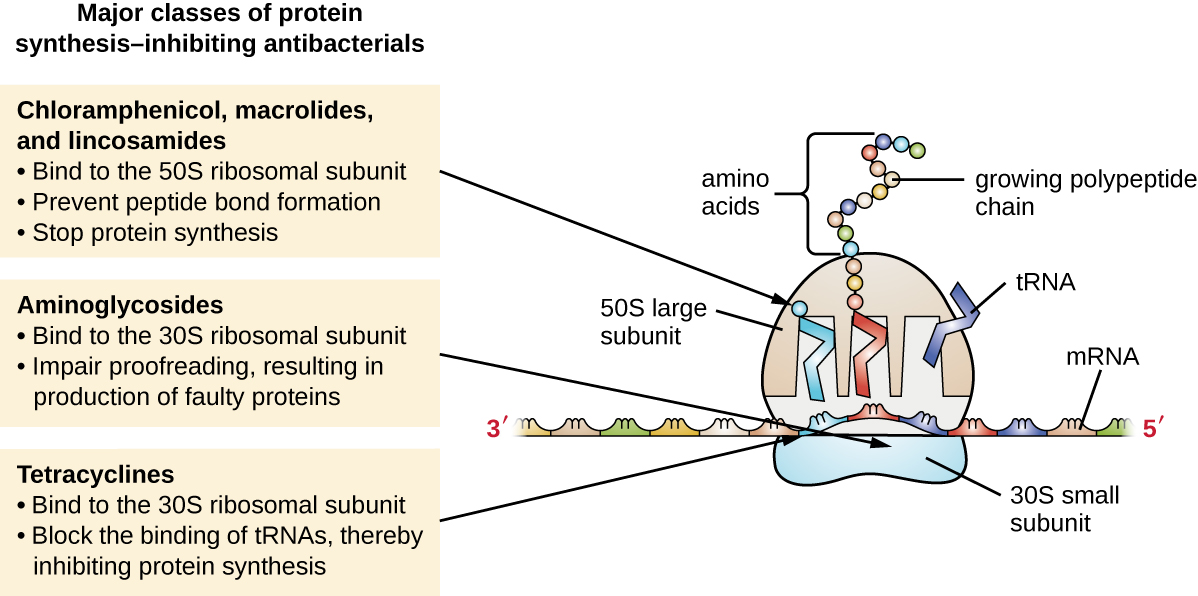
动物细胞(80S)中发现的细胞质核糖体在结构上与细菌细胞(70S)中发现的细胞质核糖体不同,这使得蛋白质生物合成成为抗菌药物的良好选择性靶标。 本节讨论了几种类型的蛋白质生物合成抑制剂,并在图中进行了总结\(\PageIndex{3}\)。
结合 30S 亚单位的蛋白质合成抑制剂
氨基糖苷类是大型、高度极性的抗菌药物,与细菌核糖体的 30S 亚基结合,削弱了核糖体复合物的校对能力。 这种损伤会导致密码子和反密码子之间的不匹配,从而产生氨基酸不正确的蛋白质和插入细胞质膜的蛋白质缩短。 有缺陷的蛋白质破坏细胞质膜会杀死细菌细胞。 氨基糖苷类药物包括链霉素、庆大霉素、新霉素和卡那霉素等药物,是有效的广谱抗菌药物。 但是,氨基糖苷类已被证明具有肾毒性(损害肾脏)、神经毒性(损害神经系统)和耳毒性(损坏耳朵)。
与30S亚单位结合的另一类抗菌化合物是四环素。 与氨基糖苷类相比,这些药物具有抑菌作用,通过在翻译过程中阻断tRNA与核糖体的关联来抑制蛋白质合成。 由各种链霉菌菌株产生的天然存在的四环素最早是在20世纪40年代发现的,还生产了几种半合成的四环素,包括强力霉素和替加环素。 尽管四环素对细菌病原体的覆盖范围广泛,但可能限制其使用的副作用包括光毒性、发育中的牙齿永久变色以及高剂量或肾功能受损患者的肝毒性。
结合 50S 亚单位的蛋白质合成抑制剂
有几类抗菌药物通过与细菌核糖体的50S亚单位结合起作用。 大环内酯类抗菌药物具有大而复杂的环状结构,是更大类别的天然产生的次生代谢物的一部分,称为聚酮类,聚酮是通过反复添加双碳单位逐步产生的复杂化合物,其机制与用于脂肪的机制相似酸合成。 大环内酯类是广谱抑菌药物,通过抑制特定氨基酸组合之间的肽键形成来阻断蛋白质的伸长。 第一个大环内酯是红霉素。 它于 1952 年从红斑链霉菌中分离出来,可防止易位。 半合成大环内酯类包括阿奇霉素和泰利霉素。 与红霉素相比,阿奇霉素的活性范围更广,副作用更少,半衰期明显更长(红霉素为1.5小时,阿奇霉素为68小时),允许大多数感染每天给药一次和短暂的3天疗程(即Zpac配方)。 Telithromycin 是被称为 ketolides 的类别中的第一种半合成物。 尽管泰利霉素对抗大环内酯类耐药性病原体的效力和活性有所提高,但美国食品药品监督管理局(FDA)将其使用仅限于治疗社区获得性肺炎,并且由于严重的肝毒性,要求该药物贴上最强的 “黑匣子警告” 标签。
林可酰胺包括天然产生的林可霉素和半合成克林霉素。 尽管在结构上与大环内酯不同,但林可酰胺通过与50S核糖体亚单位结合并防止肽键的形成,其作用方式与大环内酯类似。 Lincosamides 对链球菌和葡萄球菌感染特别活跃。
氯霉素药物是另一类结构上截然不同的抗菌药物,它们也与50S核糖体结合,抑制肽键的形成。 委内瑞拉链霉菌生产的氯霉素于1947年被发现;1949年,它成为第一个获得FDA批准的广谱抗生素。 尽管它是一种天然抗生素,但它也很容易合成,是第一种合成批量生产的抗菌药物。 由于氯霉素的批量生产、广谱覆盖范围和有效渗透到组织中的能力,历来被用于治疗从脑膜炎到伤寒再到结膜炎的各种感染。 不幸的是,严重的副作用,例如致命的灰色婴儿综合症和抑制骨髓产生,限制了其临床作用。 氯霉素还以两种不同的方式导致贫血。 一种机制涉及靶向造血干细胞中的线粒体核糖体,从而导致血细胞产生可逆的、剂量依赖性的抑制。 一旦停止服用氯霉素,血细胞产量就会恢复正常。 这种机制凸显了我们线粒体中70S的细菌核糖体和70年代的核糖体之间的相似之处。 贫血的第二种机制是特异性的(即机制尚不清楚),涉及不可逆转的致命血细胞产量损失,称为再生障碍性贫血。 再生障碍性贫血的这种机制不依赖剂量,可以在治疗停止后发展。 出于毒性考虑,人体使用氯霉素现在在美国很少见,仅限于毒性较小的抗生素无法治疗的严重感染。 由于其对动物的副作用要轻得多,因此用于兽医。
包括利奈唑胺在内的恶唑烷酮是一种新的广谱合成蛋白质合成抑制剂,与革兰氏阳性和革兰氏阴性细菌的50S核糖体亚基结合。 但是,它们的作用机制似乎与已经讨论过的其他50S亚单位结合蛋白合成抑制剂的作用机制有所不同。 相反,它们似乎干扰了起始复合物(50S亚基、30S亚基和其他因素的关联)的形成以进行翻译,并且它们阻止了生长中的蛋白质从核糖体A位点转移到P位点。 表\(\PageIndex{3}\)总结了蛋白质合成抑制剂。
| 分子靶点 | 行动机制 | 药物类别 | 特定药物 | 抑菌或杀菌 | 活动范围 |
|---|---|---|---|---|---|
| 30S 子单位 | 导致密码子和反密码子不匹配,导致蛋白质插入细胞质膜并破坏细胞质膜的缺陷 | 氨基糖苷类 | 链霉素、庆大霉素、新霉素、卡那霉素 | 杀菌 | 广谱 |
| 阻断 tRNA 与核糖体的关联 | 四环素 | 四环素、多西环素、替加环素 | 抑菌 | 广谱 | |
| 50S 子单位 | 阻断氨基酸之间肽键的形成 | 大环内酯类 | 红霉素、阿奇霉素、泰利霉素 | 抑菌 | 广谱 |
| Lincosamides | 林可霉素、克林霉素 | 抑菌 | 窄光谱 | ||
| 不适用 | 氯霉素 | 抑菌 | 广谱 | ||
| 干扰50和30S亚基和其他因素之间起始复合体的形成。 | Oxazolidinones | 利奈唑胺 | 抑菌 | 广谱 |
练习\(\PageIndex{2}\)
比较和对比不同类型的蛋白质合成抑制剂。
膜功能抑制剂
一小部分抗菌药物将细菌膜作为其作用方式(表\(\PageIndex{4}\))。 多粘菌素是天然的多肽抗生素,最初是在1947年作为多粘芽孢杆菌的产物被发现的;临床上仅使用了多粘菌素B和多粘菌素E(粘菌素)。 它们具有亲脂性,具有类似洗涤剂的特性,与革兰氏阴性细菌外膜中的脂多糖成分相互作用,最终破坏其外膜和内膜并杀死细菌细胞。 不幸的是,膜靶向机制不是选择性毒性,全身给药时,这些药物还会靶向和损害肾脏和神经系统中的细胞膜。 由于这些严重的副作用及其对消化道的吸收不良,polymyxin B 用于非处方局部抗生素软膏(例如 Neosporin),而口服粘菌素历来仅用于肠道净化,以防止肠道微生物引起的感染免疫功能低下的患者或接受某些腹部手术的患者。 但是,耐多药病原体的出现和传播导致医院越来越多地使用静脉注射粘菌素,这通常是治疗严重感染的最后手段。 抗菌达托霉素是一种由玫瑰链霉菌产生的环状脂肽,其作用看起来像多粘菌素,插入细菌细胞膜并破坏细菌细胞膜。 但是,与仅靶向革兰氏阴性细菌的多粘菌素B和大肠杆菌素相反,达托霉素专门靶向革兰氏阳性细菌。 它通常通过静脉注射,似乎耐受性良好,在骨骼肌中显示出可逆的毒性。
| 行动机制 | 药物类别 | 特定药物 | 活动范围 | 临床用途 |
|---|---|---|---|---|
| 与革兰氏阴性细菌外膜中的脂多糖相互作用,通过最终破坏外膜和细胞质膜来杀死细胞 | Polymyxins | 多粘菌素 B | 针对革兰氏阴性细菌(包括耐多药菌株)的窄光谱 | 防止伤口感染的外用制剂 |
| Polymyxin E(粘菌素) | 针对革兰氏阴性细菌(包括耐多药菌株)的窄光谱 | 口服剂量对肠道进行净化,以防止免疫功能低下患者或接受侵入性手术/手术的患者感染。 | ||
| 静脉注射治疗由耐多药病原体引起的严重全身性感染 | ||||
| 插入革兰氏阳性细菌的细胞质膜,破坏膜并杀死细胞 | Lipopeptide | 达托霉素 | 针对革兰氏阳性细菌(包括耐多药菌株)的窄光谱 | 革兰阳性病原体(包括金黄色葡萄球菌)引起的复杂皮肤和皮肤结构感染和菌血症 |
练习\(\PageIndex{3}\)
多粘菌素如何抑制膜功能?
核酸合成抑制剂
一些抗菌药物通过抑制核酸合成起作用(表\(\PageIndex{5}\))。 例如,甲硝唑是硝基咪唑家族的半合成成员,也是一种抗原生动物。 它干扰靶细胞中的 DNA 复制。 药物利福平是利福霉素家族的半合成成员,其作用是阻断细菌中的RNA聚合酶活性。 细菌中的RNA聚合酶与真核生物中的RNA聚合酶在结构上不同,从而对细菌细胞具有选择性毒性。 它用于治疗各种感染,但其主要用途(通常与其他抗菌药物混合使用)是对抗引起结核的分枝杆菌。 尽管其机制具有选择性,但利福平可以诱导肝酶增加其他正在服用的药物的新陈代谢(拮抗作用),从而导致肝毒性(肝毒性),并对伴随药物的生物利用度和治疗效果产生负面影响。
喹诺酮家族(一组合成抗微生物药物)的一个成员是萘地西酸。 它于 1962 年在合成抗疟药物氯喹时被发现为副产品。 萘地西酸选择性地抑制细菌 DNA 回旋酶的活性,阻断 DNA 复制。 对原始喹诺酮骨干的化学修饰导致了氟喹诺酮类药物的产生,例如环丙沙星和左氧氟沙星,它们也会抑制DNA回旋酶的活性。 环丙沙星和左氧氟沙星对多种革兰氏阳性或革兰氏阴性细菌均有效,是用于治疗各种感染(包括尿路感染、呼吸道感染、腹部感染和皮肤感染)的最常用处方抗生素之一。 然而,尽管它们对DNA回旋酶具有选择性毒性,但与不同的氟喹诺酮相关的副作用包括光毒性、神经毒性、心脏毒性、葡萄糖代谢功能障碍和肌腱断裂风险增加。
| 行动机制 | 药物类别 | 特定药物 | 活动范围 | 临床用途 |
|---|---|---|---|---|
| 抑制细菌 RNA 聚合酶活性并阻断转录,杀死细胞 | 利福霉素 | 利福平 | 具有抗革兰氏阳性活性的窄谱和有限数量的革兰阴性细菌。 对结核分枝杆菌也有活性。 | 治疗结核病的联合疗法 |
| 抑制 DNA 回旋酶活性并阻断 DNA 复制,杀死细胞 | 氟喹诺酮类 | 环丙沙星、氧氟沙星、莫西沙星 | 广谱抗革兰氏阳性和革兰氏阴性细菌 | 各种各样的皮肤和全身感染 |
练习\(\PageIndex{4}\)
为什么细菌核酸合成抑制剂不能靶向宿主细胞?
代谢途径抑制剂
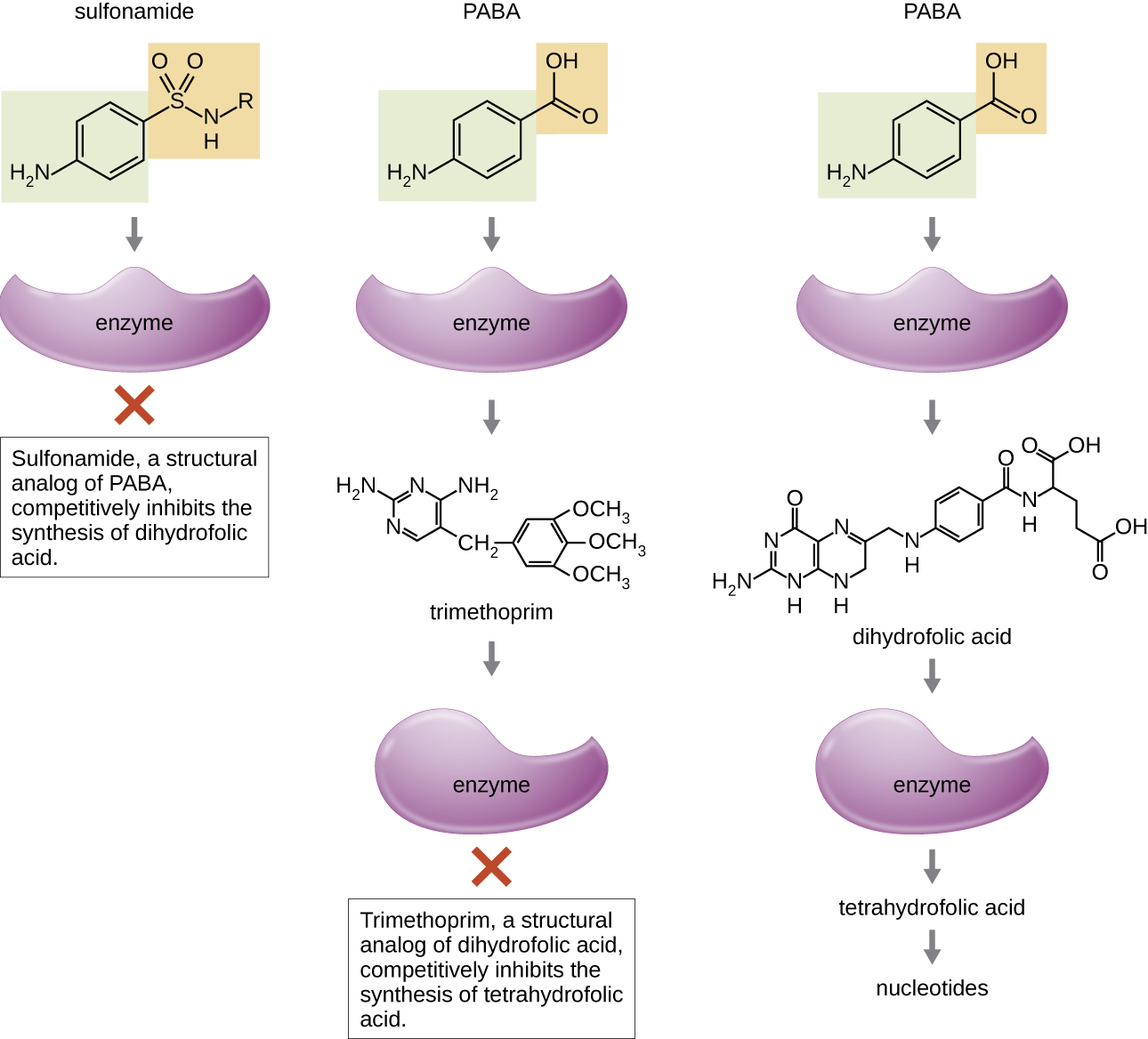
一些合成药物通过充当抗代谢物(细菌代谢酶的竞争抑制剂)来控制细菌感染(表\(\PageIndex{6}\))。 磺胺类药物(磺胺类药物)是最古老的合成抗菌剂,是叶酸合成的早期中间体对氨基苯甲酸(PABA)的结构类似物(图\(\PageIndex{4}\))。 通过抑制参与产生二氢叶酸的酶,磺胺类药物会阻断叶酸的细菌生物合成,进而阻断核酸合成所需的嘧啶和嘌呤的生物合成。 这种作用机制可抑制各种革兰氏阳性和革兰氏阴性病原体的生长。 由于人类从食物中获取叶酸而不是在细胞内合成叶酸,因此磺胺类药物对细菌具有选择性毒性。 但是,对磺胺类药物的过敏反应很常见。 磺酮在结构上与磺胺类药物相似,但除了用于治疗汉森氏病(麻风病)外,目前并不常用。
甲氧苄啶是一种合成抗菌化合物,在与磺胺类药物相同的叶酸合成途径中用作抗代谢物。 但是,甲氧苄啶是二氢叶酸的结构类似物,可抑制代谢途径的后期步骤(图\(\PageIndex{4}\))。 甲氧苄啶与磺胺类药物磺胺甲恶唑联合使用,用于治疗尿路感染、耳部感染和支气管炎。 如前所述,甲氧苄啶和磺胺甲恶唑的组合是抗菌协同作用的一个例子。 单独使用时,每种抗代谢药只会将叶酸的产生减少到抑菌抑制生长的水平。 但是,当联合使用时,抑制代谢途径中的两个步骤会使叶酸合成降低到对细菌细胞致命的水平。 由于叶酸在胎儿发育中的重要性,在妊娠初期,应仔细考虑磺胺类药物和甲氧苄啶的使用。
异烟肼是一种对分枝杆菌具有特异毒性的抗代谢药物,长期以来一直与利福平或链霉素联合用于治疗结核病。 它作为前药给药,需要通过细胞内细菌过氧化物酶的作用进行激活,形成异烟肼-烟酰胺腺嘌呤二核苷酸(NAD)和异烟肼-烟酰胺腺嘌呤磷酸二核苷酸(NADP),最终阻止霉醇酸的合成,即对分枝杆菌细胞壁至关重要。 使用异烟肼可能产生的副作用包括肝毒性、神经毒性和血液学毒性(贫血)。
| 代谢途径靶标 | 行动机制 | 药物类别 | 特定药物 | 活动范围 |
|---|---|---|---|---|
| 叶酸合成 | 抑制参与产生二氢叶酸的酶 | 磺胺类药物 | 磺胺甲恶唑 | 广谱抗革兰氏阳性和革兰氏阴性细菌 |
| 砜 | 氨苯砜 | |||
| 抑制参与产生四氢叶酸的酶 | 不适用 | 甲氧苄氨嘧啶 | 广谱抗革兰氏阳性和革兰氏阴性细菌 | |
| 霉醇酸合成 | 干扰霉醇酸的合成 | 不适用 | Isoniazid | 针对分枝杆菌属(包括结核杆菌)的窄光谱 |
练习\(\PageIndex{5}\)
磺胺类药物和甲氧苄啶如何选择性靶向细菌?
ATP 合成酶抑制剂
贝达奎林代表一种称为二芳基喹诺酮类的合成抗菌化合物,它使用一种特异性抑制分枝杆菌生长的新型作用方式。 尽管具体机制尚待阐明,但这种化合物似乎干扰了ATP合成酶的功能,可能是通过氧化磷酸化干扰氢离子梯度合成ATP,从而减少了ATP的产生。 由于其副作用,包括肝毒性和潜在的致命性心律失常,因此只能用于严重的、本来无法治愈的结核病例。
临床重点:第 2 部分
医生仔细阅读了玛丽莎的健康史,发现她在越南住院期间,接受了导管检查并接受了抗菌药物头孢他啶和甲硝唑。 得知这一点后,医生下令对玛丽莎的腹部进行 CT 扫描,以排除阑尾炎;医生还要求进行血液检查以查看她的白细胞计数是否升高,并下令进行尿液分析测试和尿液培养,以寻找是否存在白细胞、红细胞和细菌。
玛丽莎的尿液样本呈细菌存在阳性,表明存在尿路感染(UTI)。 医生开了环丙沙星处方。 同时,她的尿液经过培养以培养细菌以进行进一步检测。
练习\(\PageIndex{6}\)
- 通常为尿路感染开什么类型的抗微生物药物?
- 根据她在越南服用的抗微生物药物,你预测哪种用于治疗尿路感染的抗微生物药物无效?
关键概念和摘要
- 抗菌化合物表现出选择性毒性,这主要是由于原核细胞和真核细胞结构之间的差异。
- 细胞壁合成抑制剂,包括 β-内酰胺、糖肽和杆菌肽会干扰肽聚糖的合成,使细菌细胞更容易发生渗透裂解。
- 有多种广谱细菌蛋白合成抑制剂可以选择性靶向原核生物 70 S 核糖体,包括与 30S 亚单位(氨基糖苷类和四环素)结合的抑制剂和其他与 50S 亚基结合的抑制剂(大环内酯类、lincosamides、氯霉素和 o xazolidinones)。
- P@@ olymyxins 是亲脂性多肽抗生素,靶向革兰氏阴性细菌的脂多糖成分,最终破坏这些细菌外膜和内膜的完整性。
- 核酸合成抑制剂利福霉素和氟喹诺酮分别靶向细菌 RNA 转录和 DNA 复制。
- 一些抗菌药物是抗代谢药,充当细菌代谢酶的竞争抑制剂。 磺胺类药物和甲氧苄氨嘧啶是干扰细菌叶酸合成的抗代谢物。 异烟肼是一种抗代谢物,会干扰分枝杆菌中的霉酸合成。