
6.2: תהליך פיתוח התרופות
- Page ID
- 208254

סקירה כללית
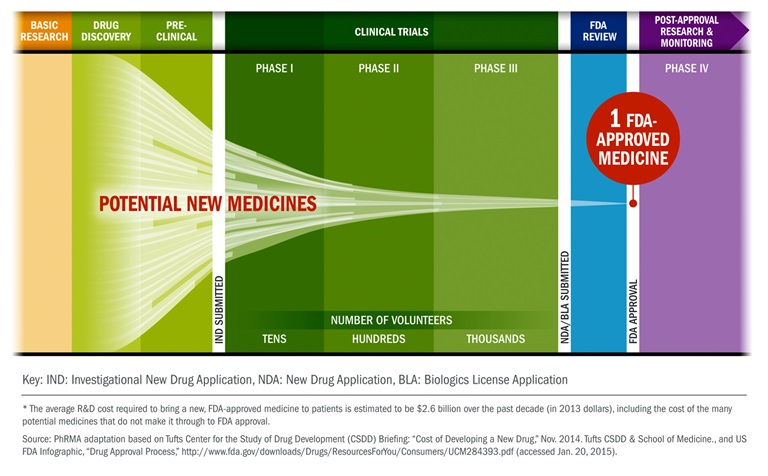
תרופות חדשות יכולות להימשך למעלה מ -12 שנים ועולות 2.6 מיליארד דולר (Phrma, 2015). אבן הדרך הראשונה לכל תרופה חדשה מתרחשת בשלב המחקר והגילוי. צורה כלשהי של ניסויים במעבדת מו"פ מובילה לפיתוח מרכיב תרופתי פעיל (API) שעשוי להיות בעל פעילות טיפולית בגוף האדם. אם מאמינים כי לחומר הפעיל יש פוטנציאל אמיתי, התרופה עוברת לשלב ההתפתחות. שלב פיתוח מוקדם זה כולל מה שמכונה "בדיקות פרה-קליניות", ומתבצע במעבדות ובמתקני ניסויים בבעלי חיים. שלב פרה-קליני זה חייב להיות מוצלח לפני שהבדיקה יכולה להתרחש בבני אדם. אם התרופה מתפקדת היטב בשלב זה, החברה יכולה להגיש בקשה לתרופות חדשות לחקירה (IND) ל- FDA כדי לבקש אישור להתחיל בבדיקות על נבדקים אנושיים. ישנם שלושה שלבים עיקריים שתרופה חייבת לעבור במהלך בדיקות נושא אנושי (מחקרים קליניים). אם התרופה עוברת מחקרים קליניים בהצלחה, החברה יכולה להגיש בקשה חדשה לתרופות (NDA) ל- FDA. אם ה- FDA יעניק את אישורו, החברה יכולה סוף סוף להתחיל למכור את המוצר, אך מעורבותו לא מסתיימת בכך. ה- FDA ימשיך לקיים אינטראקציה עם החברה כדי להבטיח שהמוצר מיוצר בבטחה. חשוב לציין כי החברה המממנת חסות לפיתוח תרופות עשויה שלא להיות החברה המבצעת את משימות הפיתוח. ארגוני מחקר חוזים (CROs) מתגייסים לעתים קרובות ומשולמים לביצוע משימות ספציפיות של פיתוח תרופות. משימות אלה עשויות לכלול ניסויים בבעלי חיים, ניסויים בבני אדם וייצור בפועל.
חקור!
ה- FDA מספק סקירה פשוטה של תהליך סקירת התרופות כאן.
האם תרופות ללא מרשם עוברות את אותו תהליך אישור?
אבני דרך בייצור תרופה
מחקר ופיתוח
הגישות המוקדמות ביותר לגילוי תרופות כללו זיהוי ובידוד המרכיבים הפעילים של כימיקלים טבעיים, כגון אלה המצויים בצמחים ובתרופות הומיאופתיות אחרות. במעבדה המודרנית, חוקרים מחפשים גם את הגורמים הסיבתיים למחלות (למשל חלבונים חסרים או פעילים יתר) כדי להנחות את מחקר התרופות על ידי הבנה לאילו בעיות יש למקד. בדיקת תפוקה גבוהה (HTS) היא גישה נוספת, המאפשרת למדענים לסנן אלפי תרופות פוטנציאליות בבת אחת ומאפשרת זיהוי ומיקוד מהירים של תרכובות שעלולות להיות שימושיות.
פיתוח פרה-קליני
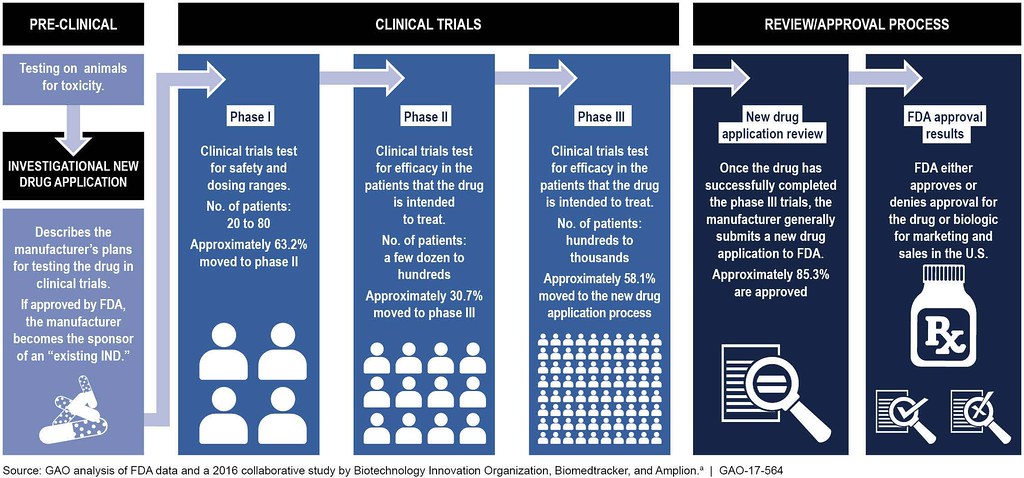
לפני בדיקת תרופה בבני אדם, היא חייבת לעבור בדיקות לא קליניות כדי להשיג רעילות בסיסית ונתונים פרמקולוגיים. בדיקות לא קליניות חייבות לכלול מודלים ומבחנים של בעלי חיים כדי לחקור פרמקולוגיה, רעילות, רעילות רבייה וגנוטוקסיות. התפתחות פרה-קלינית יכולה להימשך בין 1-4 שנים ועשויה לדרוש בדיקות נוספות כדי להיות בשילוב (או במקביל) עם מחקרים קליניים.
מטרות הפיתוח הפרה-קליני:
- זיהוי התכונות הפיזיקליות והכימיות של התרופה המועמדת
- בדיקת התרופה המועמדת במבחנה
- קביעת ניסוח למתן לנבדקים ולמטופלים
- פיתוח שיטות ייצור לתרופה המועמדת
- בדיקת התרופה המועמדת בתאים מתורבתים
- בדיקת התרופה המועמדת בבעלי חיים לבטיחות
- פיתוח מבחנים אנליטיים
- אבטחת הגנה על קניין רוחני עבור המוצר הפוטנציאלי, השימושים בו וייצורו
אדמה
כדי שתרופה פוטנציאלית זו תהיה שימושית, עליה להיות יציבה, בטוחה ומיוצרת באופן מעשי. שלב זה מוקדש גם לקביעת פעילות התרופה, תכונותיה הכימיות והמסיסות ומתווה תוכניות ייצור כדי להבטיח את הפוטנציאל שלה כתרופה. אם התרופה מראה פוטנציאל במעבדה, השלב הבא דורש בדיקות רעילות. בדיקות אלו ידועות גם בשם מחקרי ADME (קליטה, הפצה, מטבולית והפרשה). מחקרי ADME מבוצעים על בעלי חיים ומסייעים לחוקרים לקבוע:
- כמה מהתרופה נספגת בדם?
- כיצד משתנה החומר באופן מטבולי בגוף?
- מהן השפעות הרעילות של תוצרי לוואי מטבוליים?
- כמה מהר יופרשו התרופה ותוצרי הלוואי שלה?
רעילות
הערכת בטיחות נעשית באמצעות מחקרי רעילות. מחקרים אלה נערכים באמצעות הנחיות GLP למשך 30-90 יום, במינימום שני מיני יונקים, שאחד מהם חייב להיות לא מכרסם. המינון, אורך המחקר ומורכבות המחקר קשורים למחקר הקליני המוצע; משך ומורכבות צריכים להיות שווים או עולים על מה שמוצע בבני אדם. בנוסף, אם התרופה החדשה היא גם ישות כימית חדשה (NCE) ואין לה נתונים אנושיים ארוכי טווח כלל, המחקר עשוי להידרש לעלות על 12 חודשים.
- רעילות רבייה. פוריות והתפתחות עוברית נחקרים בהרחבה גם בניסויים קליניים בבני אדם. זה כולל התפתחות עוברית מוקדמת, התפתחות עוברית עוברית, כמו גם התפתחות לפני ואחרי הלידה.
- גנוטוקסיות. גנוטוקסיות, הנטייה לפגוע במידע גנטי, נחקרת בהרחבה גם במבחנה וגם in vivo. הערכה זו של מוטגניות נבדקת הן בחיידקים והן בתאי יונקים.
- קרצינוגניות. מחקרים קרצינוגניים אינם נדרשים לפני תחילת המחקרים הקליניים וייתכן שלא יהיה צורך לבצע אותם עבור מוצרים מסוימים. מחקרים אלה עשויים להימשך למעלה מ 2 שנים כדי להשלים.
יישום תרופות חדשות לחקירה (IND)
אם המועמד לתרופה מבטיח בבדיקות הפרה-קליניות, החברה אוספת את הנתונים שלה ומגישה תוכנית לבדיקת התרופה על נבדקים אנושיים ל- FDA, הנקראת יישום התרופות החדש לחקירה (IND). ה- IND מכיל מידע ממחקרים בבעלי חיים, מידע הנוגע להרכב התרופה וייצורו ותוכנית החקירה. יישום ה- IND כולל תיאור של המוצר, תוצאות ניסויים בבעלי חיים ותוכניות להמשך בדיקות. לאחר מכן מחליט ה- FDA אם חומרי החברה מלאים מספיק כדי שהחברה תוכל להתחיל לבדוק את המוצר בבני אדם.
ה- IND אינו 'מאושר'; במקום זאת, הוא הופך לפעיל תוך 30 יום מרגע קבלת ה- FDA. אם יתגלו ליקויים, ניתנת לחברה הזדמנות לתקן זאת. אם הנושאים לא יטופלו, ה- FDA יעכב את המחקרים הקליניים עד שהם יהיו. כמה תחומי דאגה ל- FDA כוללים סיכון בלתי סביר לבריאות האדם, חוקרים ללא אישורים מתאימים ונתונים פרה-קליניים לא שלמים (או מטעים).
תיקוני IND
במהלך הפיתוח הקליני, יש לעדכן את ה- IND אם יבוצעו שינויים כלשהם. תיקונים אלה עשויים לכלול שינויים בפרוטוקולים, נתוני טוקסיקולוגיה חדשים ממחקרים בבעלי חיים שהתרחבו במחקרים הקליניים, כל תופעות לוואי וכל ממצא חדש החושף תרופה זו עלול לגרום לסיכון בריאותי משמעותי למתנדבים אנושיים.
פיתוח קליני
לממשלה יש אינטרס להגן על הציבור מפני מוצרים וסמים פגומים. לכן על חברות להפגין את יעילותן ובטיחותן לפני הפצה המונית. עם זאת, הדרך היחידה שהם יכולים לעשות זאת באופן פעיל היא על ידי כך שנבדקים אנושיים יבדקו את המוצרים שלהם. כרענון מהפרק הקודם על מחקרים קליניים, צפה בסרטון זה על ניסויים קליניים: Youtu.be/PM1IGF85UOA
בנק נתונים לניסויים קליניים
כפי שנדון בעבר, כל הנתונים הקליניים מתפרסמים בבנק הנתונים של הניסויים הקליניים בכתובת clinicaltrials.gov, המתוחזק על ידי המכונים הלאומיים לבריאות (NIH). חברות נדרשות להגיש את הנתונים הקליניים שלב 2 ו -3 שלהן. חברה רשאית לבקש למנוע נתונים אלה אם הם יכולים להוכיח שהם יפריעו באופן משמעותי למחקר קליני בזמן; עם זאת, זה תלוי ב- FDA.