
12.2 : Visualisation et caractérisation de l'ADN
- Page ID
- 187899

Objectifs d'apprentissage
- Expliquer l'utilisation de sondes d'acides nucléiques pour visualiser des séquences d'ADN spécifiques
- Expliquer l'utilisation de l'électrophorèse sur gel pour séparer des fragments d'ADN
- Expliquer le principe de l'analyse du polymorphisme de longueur des fragments de restriction et ses utilisations.
- Comparez et contrastez les taches sud et nord
- Expliquer les principes et les utilisations de l'analyse par micropuces
- Décrire les méthodes utilisées pour séparer et visualiser les variants protéiques
- Expliquer la méthode et les utilisations de la réaction en chaîne par polymérase et du séquençage de
La séquence d'une molécule d'ADN peut nous aider à identifier un organisme par rapport aux séquences connues conservées dans une base de données. La séquence peut également nous renseigner sur la fonction d'une partie particulière de l'ADN, par exemple si elle code pour une protéine particulière. La comparaison des signatures protéiques, c'est-à-dire les niveaux d'expression de réseaux spécifiques de protéines, entre des échantillons est une méthode importante pour évaluer les réponses cellulaires à une multitude de facteurs et de stress environnementaux. L'analyse des signatures protéiques peut révéler l'identité d'un organisme ou la façon dont une cellule réagit en cas de maladie.
L'ADN et les protéines d'intérêt sont microscopiques et sont généralement mélangés à de nombreuses autres molécules, y compris de l'ADN ou des protéines sans rapport avec nos intérêts. De nombreuses techniques ont été développées pour isoler et caractériser les molécules d'intérêt. Ces méthodes ont été développées à l'origine à des fins de recherche, mais dans de nombreux cas, elles ont été simplifiées au point de permettre une utilisation clinique de routine. Par exemple, de nombreux agents pathogènes, tels que la bactérie Helicobacter pylori, responsable des ulcères d'estomac, peuvent être détectés à l'aide de tests à base de protéines. En outre, un nombre croissant de tests d'identification hautement spécifiques et précis basés sur l'amplification de l'ADN peuvent désormais détecter des agents pathogènes tels que les bactéries entériques résistantes aux antibiotiques, le virus de l'herpès simplex, le virus de la varicelle et du zona et bien d'autres.
Analyse moléculaire de l'ADN
Dans cette sous-section, nous décrirons certaines des méthodes de base utilisées pour séparer et visualiser des fragments spécifiques d'ADN qui intéressent un scientifique. Certaines de ces méthodes ne nécessitent pas de connaître la séquence complète de la molécule d'ADN. Avant l'avènement du séquençage rapide de l'ADN, ces méthodes étaient les seules disponibles pour fonctionner avec l'ADN, mais elles constituent toujours l'arsenal de base des outils utilisés par les généticiens moléculaires pour étudier les réponses de l'organisme aux maladies microbiennes et autres.
Sondage des acides nucléiques
Les molécules d'ADN sont petites et les informations contenues dans leur séquence sont invisibles. Comment un chercheur peut-il isoler une partie particulière de l'ADN ou, après l'avoir isolée, déterminer de quel organisme elle provient, quelle est sa séquence ou quelle est sa fonction ? L'une des méthodes permettant d'identifier la présence d'une certaine séquence d'ADN utilise des fragments d'ADN fabriqués artificiellement appelés sondes. Les sondes peuvent être utilisées pour identifier différentes espèces bactériennes dans l'environnement et de nombreuses sondes d'ADN sont désormais disponibles pour détecter les agents pathogènes de manière clinique. Par exemple, des sondes d'ADN sont utilisées pour détecter les agents pathogènes vaginaux Candida albicans, Gardnerella vaginalis et Trichomonas vaginalis.
Pour sélectionner un gène ou une séquence d'intérêt dans une bibliothèque génomique, les chercheurs doivent en savoir plus sur ce gène. Si les chercheurs possèdent une partie de la séquence d'ADN du gène d'intérêt, ils peuvent concevoir une sonde à ADN, un fragment d'ADN monocaténaire complémentaire à une partie du gène d'intérêt et différent des autres séquences d'ADN présentes dans l'échantillon. La sonde d'ADN peut être synthétisée chimiquement par des laboratoires commerciaux, ou elle peut être créée par clonage, isolement et dénaturation d'un fragment d'ADN d'un organisme vivant. Dans les deux cas, la sonde d'ADN doit être marquée à l'aide d'une étiquette moléculaire ou d'une balise, telle qu'un atome de phosphore radioactif (comme c'est le cas pour l'autoradiographie) ou un colorant fluorescent (comme c'est le cas pour l'hybridation in situ fluorescente, ou FISH), afin que la sonde et l'ADN auquel elle se lie soient visibles (Figure \(\PageIndex{1}\)). L'échantillon d'ADN à sonder doit également être dénaturé pour le rendre monocaténaire afin que la sonde d'ADN monocaténaire puisse recuire jusqu'à l'échantillon d'ADN monocaténaire aux endroits où leurs séquences sont complémentaires. Bien que ces techniques soient utiles pour le diagnostic, leur utilisation directe sur les expectorations et autres échantillons corporels peut être problématique en raison de la nature complexe de ces échantillons. L'ADN doit souvent être isolé d'échantillons corporels par des méthodes d'extraction chimique avant de pouvoir utiliser une sonde à ADN pour identifier les agents pathogènes.
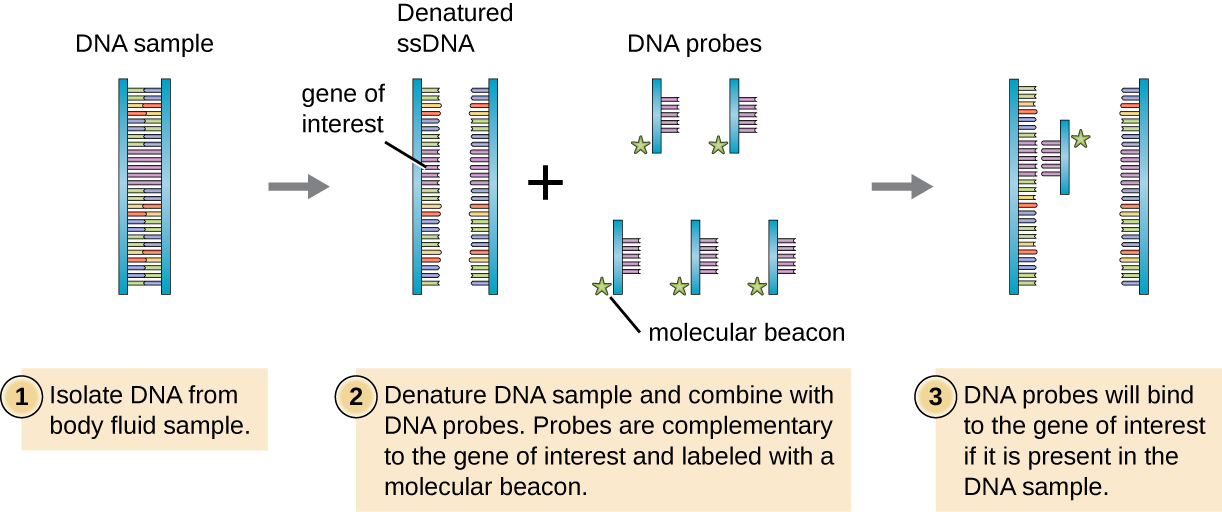
Orientation clinique : partie 2
Les symptômes légers de type grippal que ressent Kayla peuvent être causés par un certain nombre d'agents infectieux. De plus, plusieurs maladies auto-immunes non infectieuses, telles que la sclérose en plaques, le lupus érythémateux disséminé (LED) et la sclérose latérale amyotrophique (SLA), présentent également des symptômes qui correspondent aux premiers symptômes de Kayla. Cependant, au fil des semaines, les symptômes de Kayla se sont aggravés. Elle a commencé à ressentir des douleurs articulaires aux genoux, des palpitations cardiaques et une étrange faiblesse des muscles faciaux. De plus, elle souffrait d'une raideur de la nuque et de maux de tête douloureux. À contrecœur, elle a décidé qu'il était temps de consulter un médecin.
Exercice\(\PageIndex{1}\)
- Les nouveaux symptômes de Kayla fournissent-ils des indices sur le type d'infection ou d'autre problème de santé qu'elle pourrait avoir ?
- Quels tests ou outils un professionnel de santé peut-il utiliser pour identifier l'agent pathogène à l'origine des symptômes de Kayla ?
Électrophorèse sur gel d'agarose
Il existe un certain nombre de situations dans lesquelles un chercheur peut souhaiter séparer physiquement une collection de fragments d'ADN de différentes tailles. Un chercheur peut également digérer un échantillon d'ADN avec une enzyme de restriction pour former des fragments. La taille et le schéma de distribution des fragments qui en résultent peuvent souvent fournir des informations utiles sur la séquence des bases d'ADN qui peuvent être utilisées, un peu comme un scanner par code-barres, pour identifier l'individu ou l'espèce à laquelle appartient l'ADN.
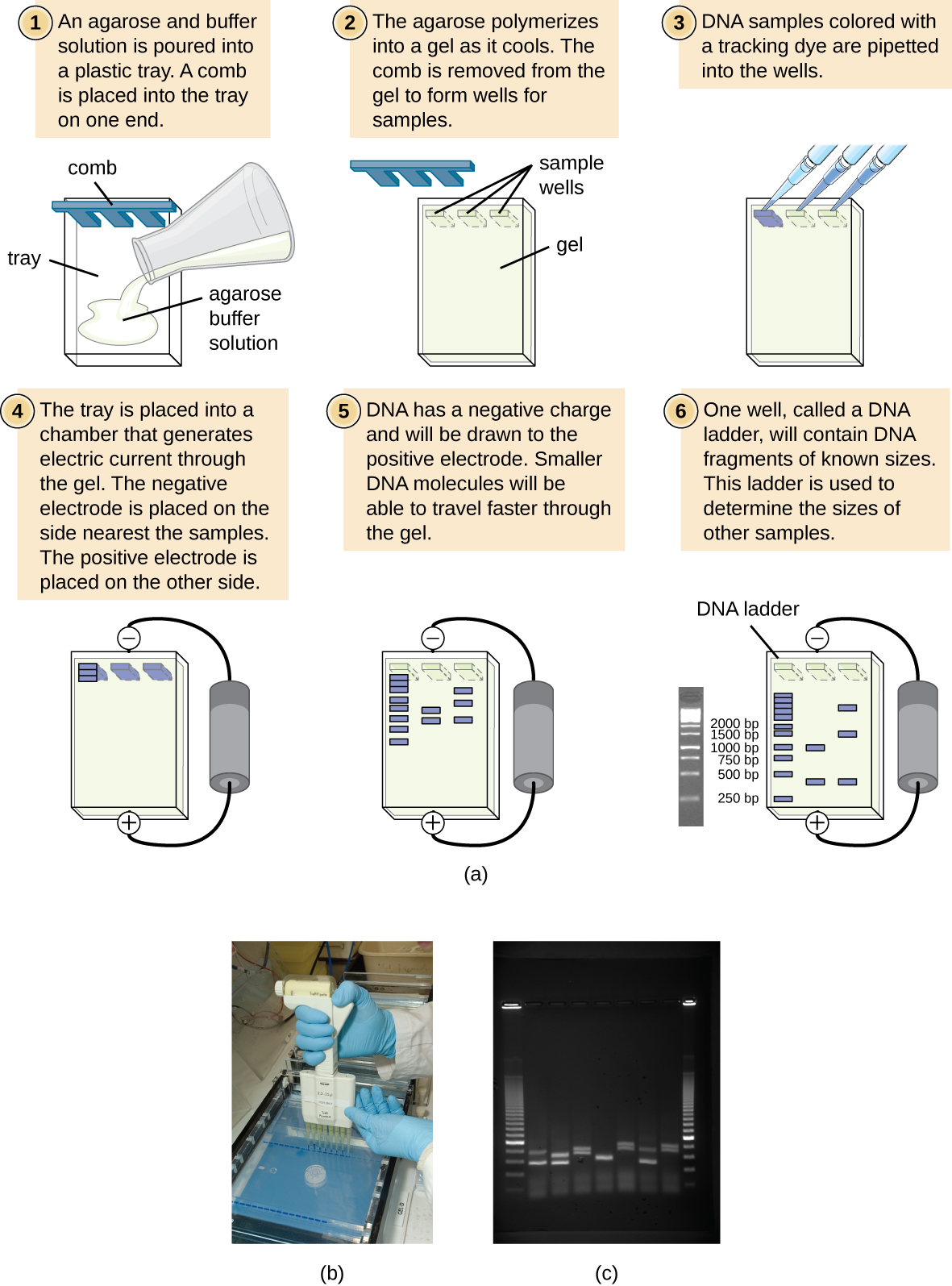
L'électrophorèse sur gel est une technique couramment utilisée pour séparer les molécules biologiques en fonction de leur taille et de leurs caractéristiques biochimiques, telles que la charge et la polarité. L'électrophorèse sur gel d'agarose est largement utilisée pour séparer l'ADN (ou ARN) de différentes tailles qui peut être généré par digestion enzymatique de restriction ou par d'autres moyens, tels que la PCR (Figure\(\PageIndex{2}\)).
En raison de son squelette chargé négativement, l'ADN est fortement attiré par une électrode positive. Lors de l'électrophorèse sur gel d'agarose, le gel est orienté horizontalement dans une solution tampon. Les échantillons sont chargés dans des puits situés sur le côté du gel le plus proche de l'électrode négative, puis extraits à travers le tamis moléculaire de la matrice d'agarose en direction de l'électrode positive. La matrice d'agarose empêche le mouvement des plus grosses molécules à travers le gel, tandis que les molécules plus petites le traversent plus facilement. Ainsi, la distance de migration est inversement corrélée à la taille du fragment d'ADN, les fragments plus petits parcourant une plus grande distance à travers le gel. La taille des fragments d'ADN d'un échantillon peut être estimée en la comparant à des fragments de taille connue dans une échelle d'ADN également traitée sur le même gel. Pour séparer de très gros fragments d'ADN, tels que des chromosomes ou des génomes viraux, l'électrophorèse sur gel d'agarose peut être modifiée en alternant périodiquement l'orientation du champ électrique pendant l'électrophorèse sur gel en champ pulsé (PFGE). Dans la PFGE, les petits fragments peuvent se réorienter et migrer légèrement plus rapidement que les fragments plus grands. Cette technique peut donc servir à séparer de très gros fragments qui se déplaceraient autrement ensemble lors d'une électrophorèse standard sur gel d'agarose. Dans n'importe laquelle de ces techniques d'électrophorèse, les emplacements des fragments d'ADN ou d'ARN dans le gel peuvent être détectés par différentes méthodes. Une méthode courante consiste à ajouter du bromure d'éthidium, un colorant qui s'insère dans les acides nucléiques à des endroits non spécifiques et qui peut être visualisé lorsqu'il est exposé à la lumière ultraviolette. D'autres colorants plus sûrs que le bromure d'éthidium, un cancérogène potentiel, sont désormais disponibles.
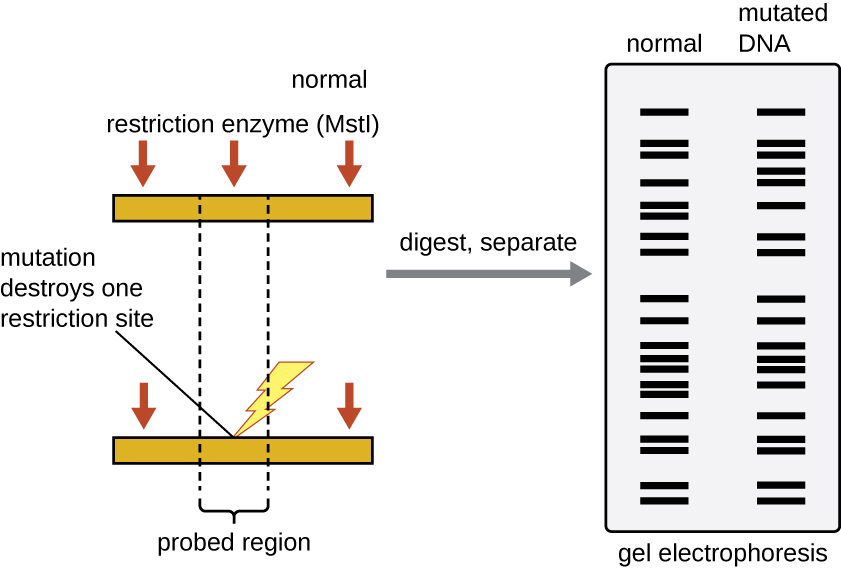
Analyse du polymorphisme de longueur des fragments de restriction (RFLP)
Les sites de reconnaissance des enzymes de restriction sont des palindromes courts (quelques nucléotides seulement), spécifiques à une séquence et peuvent être présents dans tout le génome. Ainsi, les différences dans les séquences d'ADN dans les génomes des individus entraîneront des différences dans la distribution des sites de reconnaissance des enzymes de restriction qui peuvent être visualisées sous forme de motifs de bandes distincts sur un gel après électrophorèse sur gel d'agarose. L'analyse du polymorphisme de longueur des fragments de restriction (RFLP) compare les profils de bandes d'ADN de différents échantillons d'ADN après digestion par restriction (Figure\(\PageIndex{3}\)).
L'analyse RFLP a de nombreuses applications pratiques en médecine et en criminalistique. Par exemple, les épidémiologistes utilisent l'analyse RFLP pour suivre et identifier la source de microorganismes spécifiques impliqués dans des épidémies d'intoxication alimentaire ou de certaines maladies infectieuses. L'analyse RFLP peut également être utilisée sur l'ADN humain pour déterminer les modèles d'hérédité des chromosomes dotés de gènes variants, y compris ceux associés à des maladies héréditaires ou pour établir la paternité.
Les légistes utilisent l'analyse RFLP comme forme de prise d'empreintes génétiques, ce qui est utile pour analyser l'ADN obtenu sur les scènes de crime, les suspects et les victimes. Des échantillons d'ADN sont prélevés, le nombre de copies des molécules d'ADN des échantillons est augmenté par PCR, puis soumis à une digestion par enzyme de restriction et à une électrophorèse sur gel d'agarose pour générer des motifs de bandes spécifiques. En comparant les modèles de classement des échantillons prélevés sur les lieux du crime à ceux prélevés auprès de suspects ou de victimes, les enquêteurs peuvent déterminer avec certitude si les preuves ADN collectées sur les lieux ont été laissées par les suspects ou les victimes.
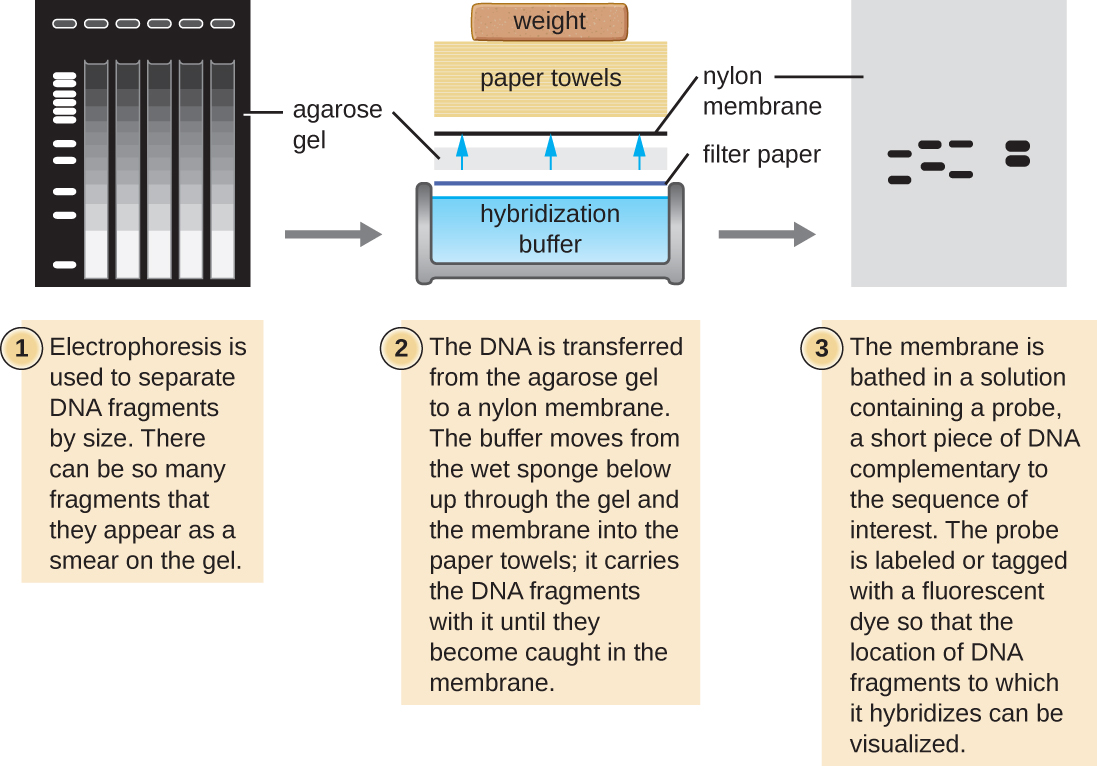
Southern Blots et modifications
Plusieurs techniques moléculaires misent sur la complémentarité des séquences et l'hybridation entre les acides nucléiques d'un échantillon et les sondes d'ADN. En général, le sondage d'échantillons d'acides nucléiques dans un gel échoue, car lorsque la sonde d'ADN pénètre dans le gel, les acides nucléiques contenus dans le gel se diffusent. Ainsi, les techniques de transfert sont couramment utilisées pour transférer des acides nucléiques vers une fine membrane chargée positivement en nitrocellulose ou en nylon. Dans la technique Southern Blot, développée par Sir Edwin Southern en 1975, les fragments d'ADN d'un échantillon sont d'abord séparés par électrophorèse sur gel d'agarose, puis transférés sur une membrane par action capillaire (Figure\(\PageIndex{4}\)). Les fragments d'ADN qui se lient à la surface de la membrane sont ensuite exposés à une sonde d'ADN monocaténaire spécifique marquée par une balise moléculaire radioactive ou fluorescente pour faciliter la détection. Les Southern blots peuvent être utilisés pour détecter la présence de certaines séquences d'ADN dans un échantillon d'ADN donné. Une fois que l'ADN cible à l'intérieur de la membrane est visualisé, les chercheurs peuvent découper la partie de la membrane contenant le fragment afin de récupérer le fragment d'ADN d'intérêt.
Les variantes du Southern blot (point blot, slot blot et spot blot) n'impliquent pas d'électrophorèse, mais concentrent plutôt l'ADN d'un échantillon à un petit endroit sur une membrane. Après hybridation avec une sonde à ADN, l'intensité du signal détecté est mesurée, ce qui permet au chercheur d'estimer la quantité d'ADN cible présente dans l'échantillon.
Le transfert de colonies est une autre variante du Southern blot dans laquelle des colonies représentant différents clones d'une bibliothèque génomique sont transférées sur une membrane en pressant la membrane sur la plaque de culture. Les cellules de la membrane sont lysées et la membrane peut ensuite être sondée pour déterminer quelles colonies d'une bibliothèque génomique abritent le gène cible. Comme les colonies de la plaque sont toujours en croissance, les cellules d'intérêt peuvent être isolées de la plaque.
Dans le Northern Blot, une autre variante du Southern blot, l'ARN (et non l'ADN) est immobilisé sur la membrane et sondé. Les Northern Blot sont généralement utilisés pour détecter la quantité d'ARNm produite par expression génique dans un échantillon de tissu ou d'organisme.
Analyse par microarray
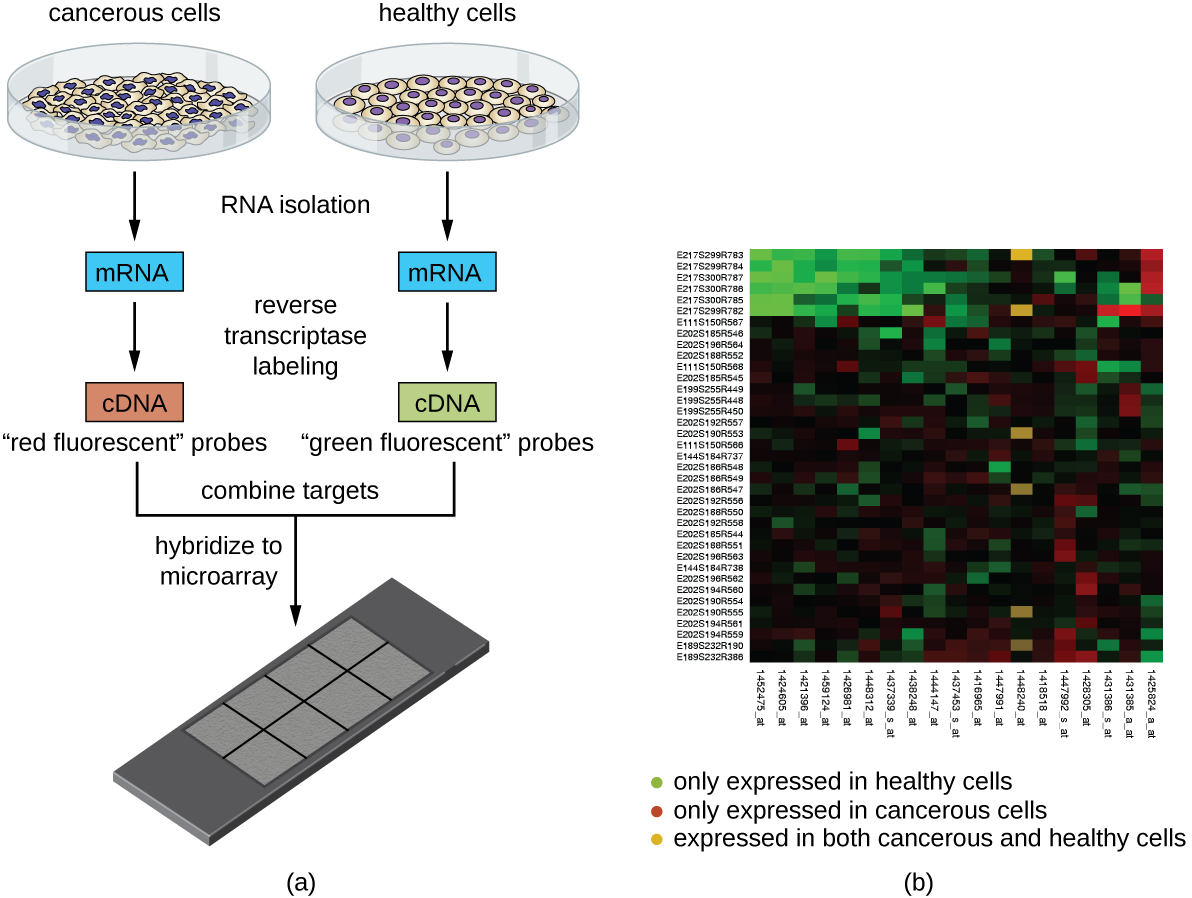
L'analyse par microréseau est une autre technique qui tire parti de l'hybridation entre des séquences d'acides nucléiques complémentaires. L'analyse par microréseau est utile pour comparer les modèles d'expression génique entre différents types de cellules, par exemple, des cellules infectées par un virus par rapport à des cellules non infectées, ou des cellules cancéreuses par rapport à des cellules saines (Figure\(\PageIndex{5}\)).
Généralement, l'ADN ou l'ADNc d'un échantillon expérimental est déposé sur une lame de verre aux côtés de séquences d'ADN connues. Chaque lame peut contenir plus de 30 000 types de fragments d'ADN différents. Des fragments d'ADN distincts (englobant l'ensemble de la bibliothèque génomique d'un organisme) ou des fragments d'ADNc (correspondant à l'ensemble complet des gènes exprimés d'un organisme) peuvent être repérés individuellement sur une lame de verre.
Une fois déposé sur la lame, l'ADN génomique ou l'ARNm peuvent être isolés des deux échantillons à des fins de comparaison. Si l'ARNm est isolé, il est retranscrit en ADNc à l'aide de la transcriptase inverse. Ensuite, les deux échantillons d'ADN génomique ou d'ADNc sont marqués avec différents colorants fluorescents (généralement rouge et vert). Les échantillons d'ADN génomique marqués sont ensuite combinés en quantités égales, ajoutés à la puce à microréseau et autorisés à s'hybrider à des points complémentaires sur le microréseau.
L'hybridation des molécules d'ADN génomique des échantillons peut être surveillée en mesurant l'intensité de la fluorescence à des endroits particuliers du microréseau. Les différences dans le degré d'hybridation entre les échantillons peuvent être facilement observées. Si les acides nucléiques d'un seul échantillon s'hybrident à un point particulier du microréseau, ce point apparaîtra vert ou rouge. Toutefois, si les acides nucléiques des deux échantillons s'hybrident, la tache apparaîtra jaune en raison de la combinaison des colorants rouge et vert.
Bien que la technologie des microréseaux permette une comparaison holistique entre deux échantillons en peu de temps, elle nécessite un équipement de détection et un logiciel d'analyse sophistiqués (et coûteux). En raison de son coût, cette technologie est généralement limitée aux environnements de recherche. Les chercheurs ont utilisé l'analyse par microréseaux pour étudier comment l'expression génique est affectée chez des organismes infectés par des bactéries ou des virus ou soumis à certains traitements chimiques.
Découvrez la technologie des micropuces sur ce site Web interactif.
Exercice\(\PageIndex{2}\)
- En quoi consiste une sonde d'ADN ?
- Pourquoi utiliser un Southern blot après l'électrophorèse sur gel d'un condensé d'ADN ?
Analyse moléculaire des protéines
Dans de nombreux cas, il peut ne pas être souhaitable ou possible d'étudier directement l'ADN ou l'ARN. Les protéines peuvent fournir des informations spécifiques à l'espèce pour l'identification ainsi que des informations importantes sur la manière dont et si une cellule ou un tissu réagit à la présence d'un microorganisme pathogène. Diverses protéines nécessitent différentes méthodes d'isolement et de caractérisation.
Électrophorèse sur gel de polyacrylamide
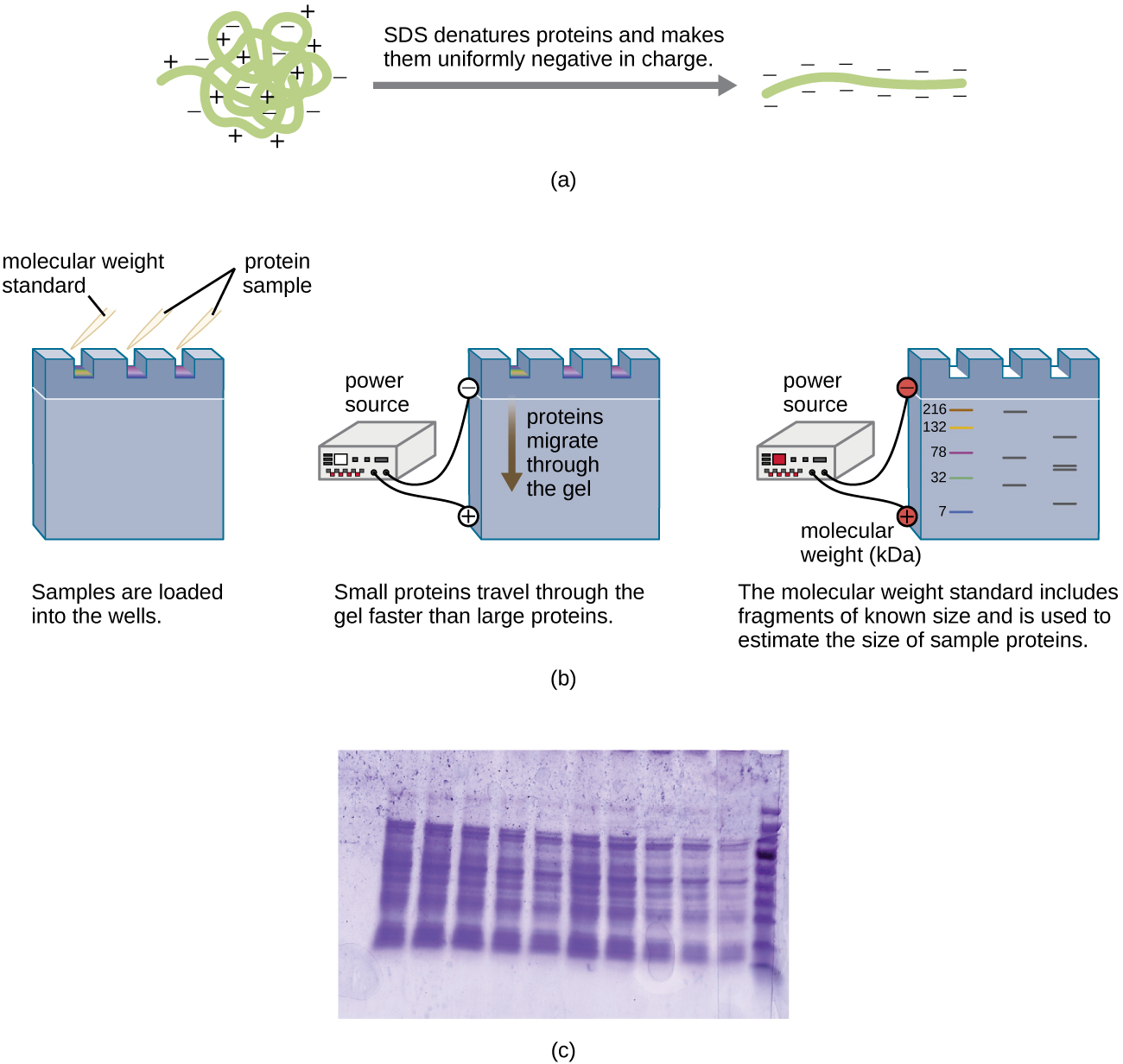
Une variante de l'électrophorèse sur gel, appelée électrophorèse sur gel de polyacrylamide (PAGE), est couramment utilisée pour séparer les protéines. Dans PAGE, la matrice du gel est plus fine et composée de polyacrylamide au lieu d'agarose. De plus, la PAGE est généralement réalisée à l'aide d'un appareil à gel vertical (Figure\(\PageIndex{6}\)). En raison des charges variables associées aux chaînes latérales des acides aminés, le PAGE peut être utilisé pour séparer les protéines intactes en fonction de leurs charges nettes. Les protéines peuvent également être dénaturées et recouvertes d'un détergent chargé négativement appelé dodécylsulfate de sodium (SDS), masquant les charges natives et permettant une séparation basée uniquement sur la taille. La PAGE peut être modifiée davantage pour séparer les protéines en fonction de deux caractéristiques, telles que leur charge à différents pH ainsi que leur taille, grâce à l'utilisation d'une PAGE bidimensionnelle. Dans tous ces cas, après électrophorèse, les protéines sont visualisées par coloration, généralement au bleu de Coomassie ou à l'argent.
Exercice\(\PageIndex{3}\)
Sur quelle base les protéines sont-elles séparées dans SDS-PAGE ?
Orientation clinique : 3e partie
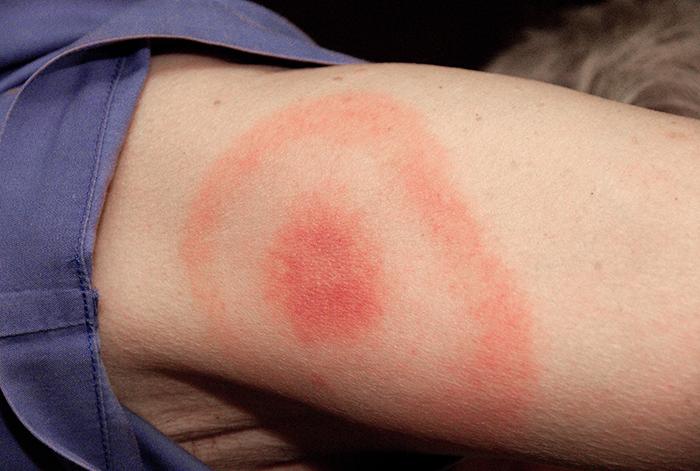
Lorsque Kayla a décrit ses symptômes, son médecin a d'abord soupçonné une méningite bactérienne, ce qui correspond à ses maux de tête et à sa raideur de la nuque. Cependant, elle a rapidement écarté cette possibilité, car la méningite progresse généralement plus rapidement que celle dont souffrait Kayla. Bon nombre de ses symptômes ressemblaient encore à ceux de la sclérose latérale amyotrophique (SLA) et du lupus érythémateux disséminé (LED), et le médecin a également considéré la maladie de Lyme comme une possibilité étant donné le temps que Kayla passe dans les bois. Kayla ne se souvenait d'aucune morsure de tique récente (le moyen typique de transmission de la maladie de Lyme) et elle ne présentait pas l'éruption cutanée typique associée à la maladie de Lyme (Figure\(\PageIndex{7}\)). Cependant, 20 à 30 % des patients atteints de la maladie de Lyme ne développent jamais cette éruption cutanée, de sorte que le médecin n'a pas voulu l'exclure.
Le médecin de Kayla a prescrit une IRM de son cerveau, une formule sanguine complète pour tester l'anémie, des analyses sanguines évaluant la fonction hépatique et rénale et des tests supplémentaires pour confirmer ou exclure le LED ou la maladie de Lyme. Les résultats de ses tests ne correspondaient pas à la fois au LED et à la SLA, et le résultat du test visant à détecter les anticorps contre la maladie de Lyme était « équivoque », c'est-à-dire non concluant. Après avoir exclu la SLA et le LE, le médecin de Kayla a décidé d'effectuer des tests supplémentaires pour détecter la maladie de Lyme.
Exercice\(\PageIndex{4}\)
- Pourquoi le médecin de Kayla soupçonne-t-il toujours la maladie de Lyme même si les résultats des tests n'ont pas détecté d'anticorps de Lyme dans le sang ?
- Quel type de test moléculaire peut être utilisé pour détecter les anticorps sanguins contre la maladie de Lyme ?
Méthodes d'analyse de l'ADN basées sur l'amplification
Diverses méthodes peuvent être utilisées pour obtenir des séquences d'ADN, qui sont utiles pour étudier les organismes pathogènes. Avec l'avènement de la technologie de séquençage rapide, notre base de connaissances sur l'ensemble du génome des organismes pathogènes s'est considérablement développée. Nous commençons par une description de la réaction en chaîne par polymérase, qui n'est pas une méthode de séquençage mais qui a permis aux chercheurs et aux cliniciens d'obtenir les grandes quantités d'ADN nécessaires au séquençage et à d'autres études. La réaction en chaîne par polymérase élimine la dépendance que nous avions autrefois à l'égard des cellules pour fabriquer de multiples copies de l'ADN, obtenant le même résultat grâce à des réactions relativement simples à l'extérieur de la cellule.
Réaction en chaîne par polymérase (PCR)
La plupart des méthodes d'analyse de l'ADN, telles que la digestion par enzymes de restriction et l'électrophorèse sur gel d'agarose, ou le séquençage de l'ADN, nécessitent de grandes quantités d'un fragment d'ADN spécifique. Dans le passé, de grandes quantités d'ADN étaient produites en cultivant les cellules hôtes d'une bibliothèque génomique. Cependant, la préparation des bibliothèques demande du temps et des efforts et les échantillons d'ADN intéressants sont souvent fournis en quantités infimes. La réaction en chaîne par polymérase (PCR) permet une amplification rapide du nombre de copies de séquences d'ADN spécifiques pour une analyse plus approfondie (Figure\(\PageIndex{8}\)). L'une des techniques les plus puissantes de biologie moléculaire, la PCR a été développée en 1983 par Kary Mullis alors qu'il travaillait pour Cetus Corporation. La PCR a des applications spécifiques dans les laboratoires de recherche, de criminalistique et clinique, notamment :
- détermination de la séquence de nucléotides dans une région spécifique de l'ADN
- amplification d'une région cible de l'ADN pour le clonage dans un vecteur plasmidique
- identifier la source d'un échantillon d'ADN laissé sur les lieux d'un crime
- analyse d'échantillons pour déterminer la paternité
- comparaison d'échantillons d'ADN ancien avec des organismes modernes
- déterminer la présence de microorganismes difficiles à cultiver ou non cultivables dans des échantillons humains ou environnementaux
La PCR est une technique de laboratoire in vitro qui tire parti du processus naturel de réplication de l'ADN. Les enzymes d'ADN polymérase thermostables utilisées dans la PCR sont dérivées de procaryotes hyperthermophiles. L'ADN polymérase Taq, couramment utilisée en PCR, est dérivée de la bactérie Thermus aquaticus isolée d'une source thermale du parc national de Yellowstone. La réplication de l'ADN nécessite l'utilisation d'amorces pour initier la réplication afin de disposer de groupes 3′-hydroxyle libres pour l'ajout de nucléotides par l'ADN polymérase. Cependant, alors que les amorces composées d'ARN sont normalement utilisées dans les cellules, les amorces d'ADN sont utilisées pour la PCR. Les amorces à ADN sont préférables en raison de leur stabilité, et les amorces d'ADN avec des séquences connues ciblant une région d'ADN spécifique peuvent être synthétisées chimiquement dans le commerce. Ces amorces d'ADN sont fonctionnellement similaires aux sondes d'ADN utilisées pour les différentes techniques d'hybridation décrites précédemment, se liant à des cibles spécifiques en raison de la complémentarité entre la séquence d'ADN cible et l'amorce.
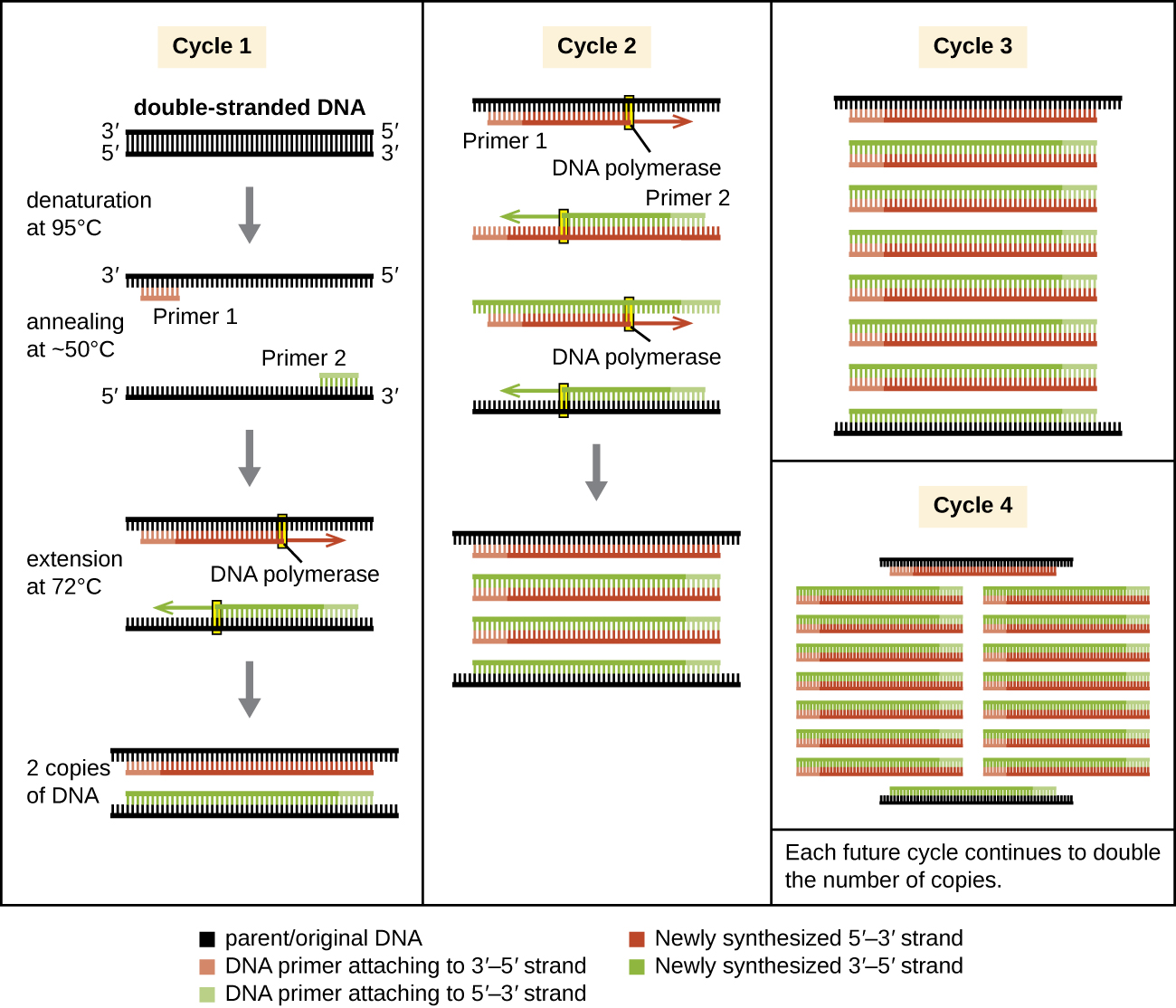
La PCR s'effectue sur plusieurs cycles, chacun comportant trois étapes : dénaturation, recuit et extension. Des machines appelées thermocycleurs sont utilisées pour la PCR ; ces machines peuvent être programmées pour passer automatiquement aux températures requises à chaque étape (Figure 12.1). Tout d'abord, l'ADN matrice double brin contenant la séquence cible est dénaturé à environ 95 °C. La température élevée requise pour séparer physiquement (plutôt qu'enzymatiquement) les brins d'ADN est la raison pour laquelle l'ADN polymérase thermostable est requise. Ensuite, la température est abaissée à environ 50 °C, ce qui permet aux amorces d'ADN complémentaires aux extrémités de la séquence cible de recuire (adhérer) aux brins de la matrice, une amorce recuite étant effectuée sur chaque brin. Enfin, la température est portée à 72 °C, la température optimale pour l'activité de l'ADN polymérase thermostable, ce qui permet d'ajouter des nucléotides à l'amorce en utilisant la cible monocaténaire comme matrice. Chaque cycle double le nombre de copies d'ADN cible bicaténaires. En général, les protocoles PCR incluent 25 à 40 cycles, ce qui permet l'amplification d'une seule séquence cible par des dizaines de millions, voire plus d'un billion.
La réplication naturelle de l'ADN est conçue pour copier l'ensemble du génome et commence sur un ou plusieurs sites d'origine. Les amorces sont construites pendant la réplication, pas avant, et ne se composent pas de quelques séquences spécifiques. La PCR cible des régions spécifiques d'un échantillon d'ADN à l'aide d'amorces spécifiques à la séquence. Ces dernières années, diverses méthodes d'amplification par PCR isotherme qui évitent le besoin de cyclage thermique ont été développées, en tirant parti des protéines accessoires qui facilitent le processus de réplication de l'ADN. À mesure que le développement de ces méthodes se poursuit et que leur utilisation se généralise dans les laboratoires de recherche, de criminalistique et clinique, les thermocycleurs peuvent devenir obsolètes.
Approfondissez votre compréhension de la réaction en chaîne de la polymérase en visionnant cette animation et en participant à un exercice interactif.
Variantes de PCR
Plusieurs modifications ultérieures apportées à la PCR augmentent encore l'utilité de cette technique. La PCR par transcriptase inverse (RT-PCR) est utilisée pour obtenir des copies d'ADN d'une molécule d'ARNm spécifique. La RT-PCR commence par l'utilisation de l'enzyme transcriptase inverse pour convertir les molécules d'ARNm en ADNc. Cet ADNc est ensuite utilisé comme matrice pour l'amplification par PCR traditionnelle. La RT-PCR permet de détecter si un gène spécifique a été exprimé dans un échantillon. Une autre application récente de la PCR est la PCR en temps réel, également connue sous le nom de PCR quantitative (qPCR). Les protocoles standard de PCR et de RT-PCR ne sont pas quantitatifs car l'un des réactifs peut devenir limitant avant la fin de tous les cycles du protocole et les échantillons ne sont analysés qu'à la fin. Comme il n'est pas possible de déterminer à quel moment, dans le protocole de PCR ou de RT-PCR, un réactif donné est devenu limitant, il n'est pas possible de savoir combien de cycles ont été achevés avant ce point, et il n'est donc pas possible de déterminer combien de molécules modèles d'origine étaient présentes dans l'échantillon au début de PCR. Dans la qPCR, cependant, l'utilisation de la fluorescence permet de surveiller l'augmentation d'une matrice bicaténaire au cours d'une réaction de PCR au fur et à mesure qu'elle se produit. Ces données cinétiques peuvent ensuite être utilisées pour quantifier la quantité de la séquence cible d'origine. L'utilisation de la qPCR ces dernières années a encore élargi les capacités de la PCR, permettant aux chercheurs de déterminer le nombre de copies d'ADN, et parfois d'organismes, présentes dans un échantillon. En milieu clinique, la qRT-PCR est utilisée pour déterminer la charge virale chez les patients séropositifs afin d'évaluer l'efficacité de leur traitement.
Séquençage ADN
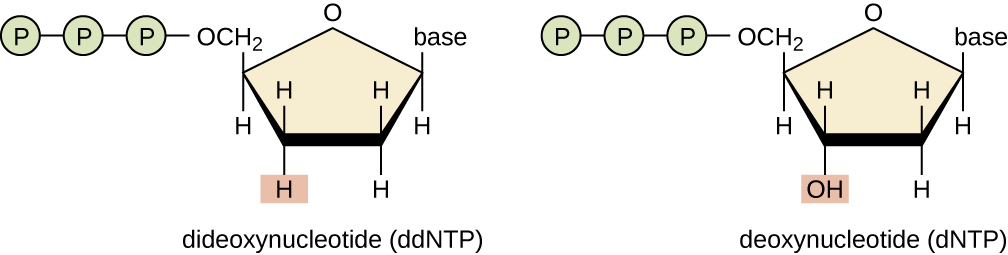
Une technique de séquençage de base est la méthode de terminaison de chaîne, également connue sous le nom de méthode didésoxy ou méthode de séquençage de l'ADN de Sanger, développée par Frederick Sanger en 1972. La méthode de terminaison de chaîne implique la réplication de l'ADN d'une matrice monocaténaire à l'aide d'une amorce d'ADN pour initier la synthèse d'un brin complémentaire, l'ADN polymérase, d'un mélange des quatre monomères réguliers de désoxynucléotides (dNTP) et d'une petite proportion de didésoxynucléotides (DDNTP), chacun étant marqué par un balise moléculaire. Les DDNTP sont des monomères dépourvus de groupe hydroxyle (—OH) au site auquel un autre nucléotide se fixe habituellement pour former une chaîne (Figure\(\PageIndex{9}\)). Chaque fois qu'un ddNTP est incorporé de manière aléatoire dans le brin complémentaire en croissance, il met fin au processus de réplication de l'ADN pour ce brin en particulier. Il en résulte de multiples brins courts d'ADN répliqué qui se terminent chacun à un point différent pendant la réplication. Lorsque le mélange réactionnel est soumis à une électrophorèse sur gel, les multiples brins d'ADN nouvellement répliqués forment une échelle de tailles différentes. Comme les DDNTP sont marqués, chaque bande du gel reflète la taille du brin d'ADN lorsque le ddNTP a mis fin à la réaction.
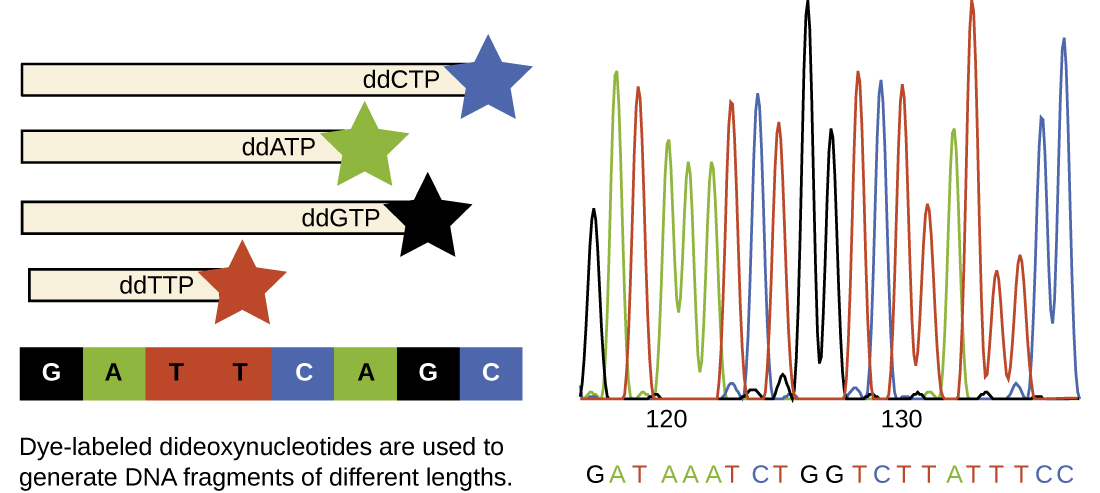
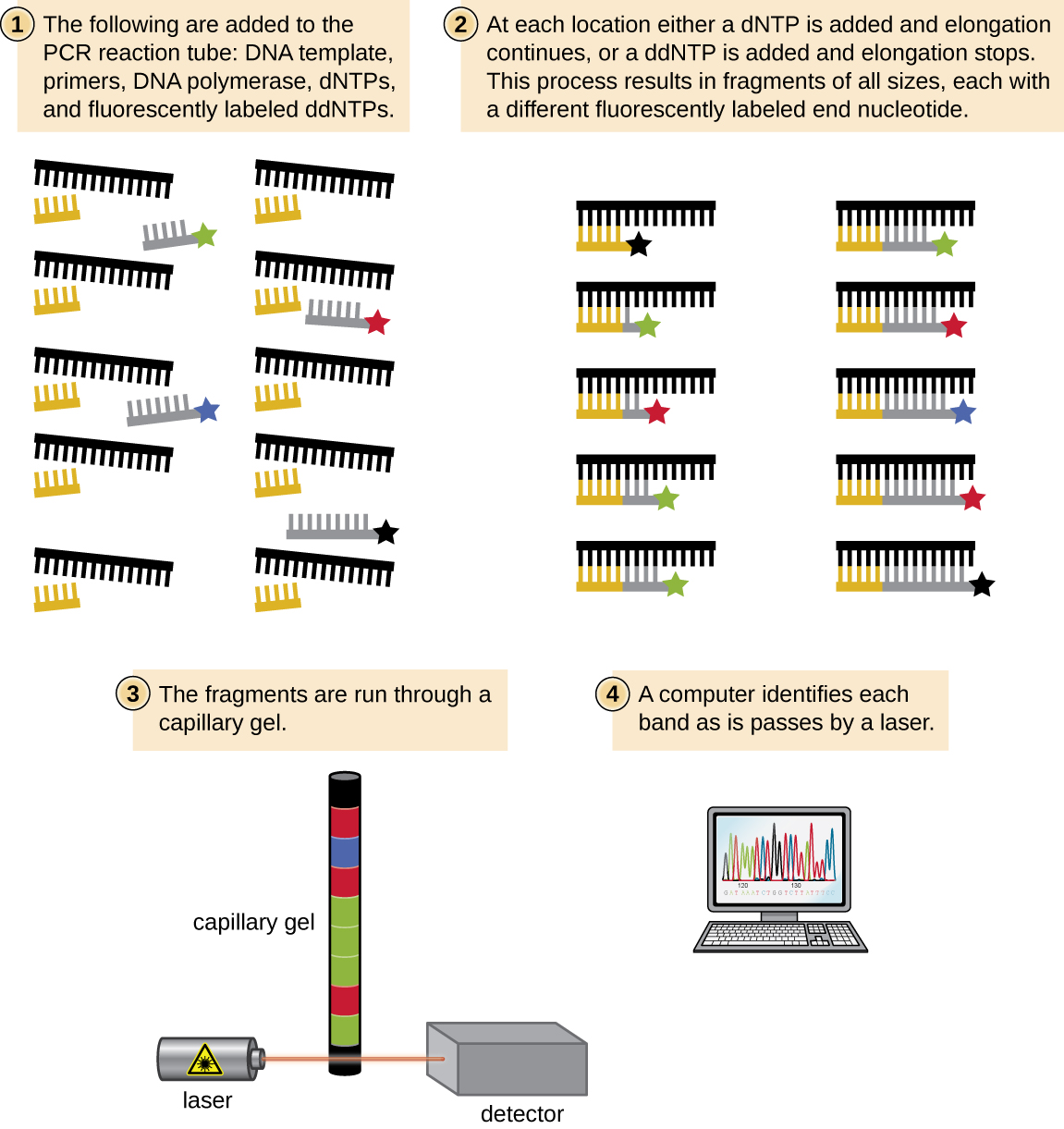
À l'époque de Sanger, quatre réactions étaient mises en place pour chaque molécule d'ADN séquencée, chaque réaction ne contenant qu'un seul des quatre DDNTP possibles. Chaque ddNTP a été marqué avec une molécule de phosphore radioactif. Les produits des quatre réactions ont ensuite été analysés dans des voies séparées côte à côte sur des gels PAGE longs et étroits, et les bandes de différentes longueurs ont été détectées par autoradiographie. Aujourd'hui, ce processus a été simplifié grâce à l'utilisation de DDNTP, chacun marqué avec un colorant fluorescent ou un fluorochrome de couleur différente (Figure\(\PageIndex{10}\)), dans une réaction de séquençage contenant les quatre DDNTP possibles pour chaque molécule d'ADN séquencée (Figure\(\PageIndex{11}\)). Ces fluorochromes sont détectés par spectroscopie de fluorescence. La détermination de la couleur de fluorescence de chaque bande lorsqu'elle passe devant le détecteur produit la séquence nucléotidique du brin matrice.
Depuis 2005, les techniques de séquençage automatisé utilisées par les laboratoires relèvent du séquençage de nouvelle génération, qui est un groupe de techniques automatisées utilisées pour le séquençage rapide de l'ADN. Ces méthodes ont révolutionné le domaine de la génétique moléculaire car les séquenceurs peu coûteux peuvent générer des séquences de centaines de milliers ou de millions de fragments courts (25 à 600 paires de bases) en une seule journée. Bien que plusieurs variantes des technologies de séquençage de nouvelle génération soient fabriquées par différentes entreprises (par exemple, le pyroséquençage de 454 Life Sciences et la technologie Solexa d'Illumina), elles permettent toutes de séquencer rapidement des millions de bases, ce qui rend le séquençage de génomes entiers relativement facile et peu coûteux, et banal. Lors du séquençage 454 (pyroséquençage), par exemple, un échantillon d'ADN est fragmenté en fragments monocaténaires de 400 à 600 pb, modifiés par l'ajout d'adaptateurs d'ADN aux deux extrémités de chaque fragment. Chaque fragment d'ADN est ensuite immobilisé sur une bille et amplifié par PCR, à l'aide d'amorces conçues pour être recuits aux adaptateurs, créant ainsi une bille contenant de nombreuses copies de ce fragment d'ADN. Chaque bille est ensuite placée dans un puits séparé contenant des enzymes de séquençage. Dans le puits, chacun des quatre nucléotides est ajouté l'un après l'autre ; lorsque chacun est incorporé, du pyrophosphate est libéré en tant que sous-produit de la polymérisation, émettant un petit flash de lumière qui est enregistré par un détecteur. Cela fournit l'ordre des nucléotides incorporés lors de la fabrication d'un nouveau brin d'ADN et constitue un exemple de séquençage de synthèse. Les séquenceurs de nouvelle génération utilisent des logiciels sophistiqués pour passer à travers le processus fastidieux de mise en ordre de tous les fragments. Dans l'ensemble, ces technologies continuent de progresser rapidement, diminuant le coût du séquençage et augmentant rapidement la disponibilité des données de séquence provenant d'une grande variété d'organismes.
Le National Center for Biotechnology Information héberge une base de données de séquences génétiques largement utilisée, appelée GenBank, où les chercheurs déposent des informations génétiques destinées à un usage public. Dès la publication des données de séquence, les chercheurs les téléchargent sur GenBank, ce qui permet aux autres chercheurs d'accéder à l'information. Cette collaboration permet aux chercheurs de comparer des informations sur les séquences d'échantillons récemment découvertes ou inconnues avec la vaste gamme de données de séquences qui existent déjà.
Visionnez une animation sur le séquençage 454 pour approfondir votre compréhension de cette méthode.
Utiliser un NAAT pour diagnostiquer une infection à C. difficile
Javier, un patient de 80 ans ayant des antécédents de maladie cardiaque, est récemment rentré de l'hôpital après avoir subi une angioplastie visant à insérer un stent dans une artère cardiaque. Afin de minimiser les risques d'infection, Javier a reçu des antibiotiques à large spectre par voie intraveineuse pendant et peu de temps après son intervention. Il a été libéré quatre jours après l'intervention, mais une semaine plus tard, il a commencé à ressentir de légères crampes abdominales et une diarrhée aqueuse plusieurs fois par jour. Il a perdu l'appétit, s'est déshydraté gravement et a développé de la fièvre. Il a également remarqué du sang dans ses selles. La femme de Javier a appelé le médecin, qui lui a demandé de l'emmener immédiatement aux urgences.
Le personnel de l'hôpital a effectué plusieurs tests et a découvert que le taux de créatinine rénale de Javier était élevé par rapport à son taux sanguin, ce qui indique que ses reins ne fonctionnaient pas bien. Les symptômes de Javier suggéraient une infection possible par Clostridium difficile, une bactérie résistante à de nombreux antibiotiques. L'hôpital a prélevé et cultivé un échantillon de selles pour déterminer la production des toxines A et B par C. difficile, mais les résultats se sont révélés négatifs. Cependant, les résultats négatifs n'ont pas suffi à exclure la possibilité d'une infection à C. difficile, car la culture de C. difficile et la détection de ses toxines caractéristiques peuvent s'avérer difficiles, en particulier dans certains types d'échantillons. Pour des raisons de sécurité, ils ont procédé à un test diagnostique d'amplification des acides nucléiques (NAAT). À l'heure actuelle, les NAAT constituent la référence absolue du diagnosticien clinique pour détecter le matériel génétique d'un agent pathogène. Dans le cas de Javier, la qPCR a été utilisée pour rechercher le gène codant pour la toxine B de C. difficile (TCDb). Lorsque l'analyse qPCR s'est révélée positive, le médecin traitant a conclu que Javier souffrait bien d'une infection à C. difficile et a immédiatement prescrit l'antibiotique vancomycine, à administrer par voie intraveineuse. L'antibiotique a éliminé l'infection et Javier s'est complètement rétabli.
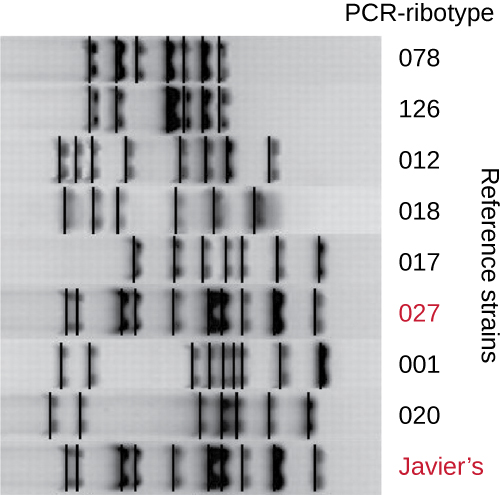
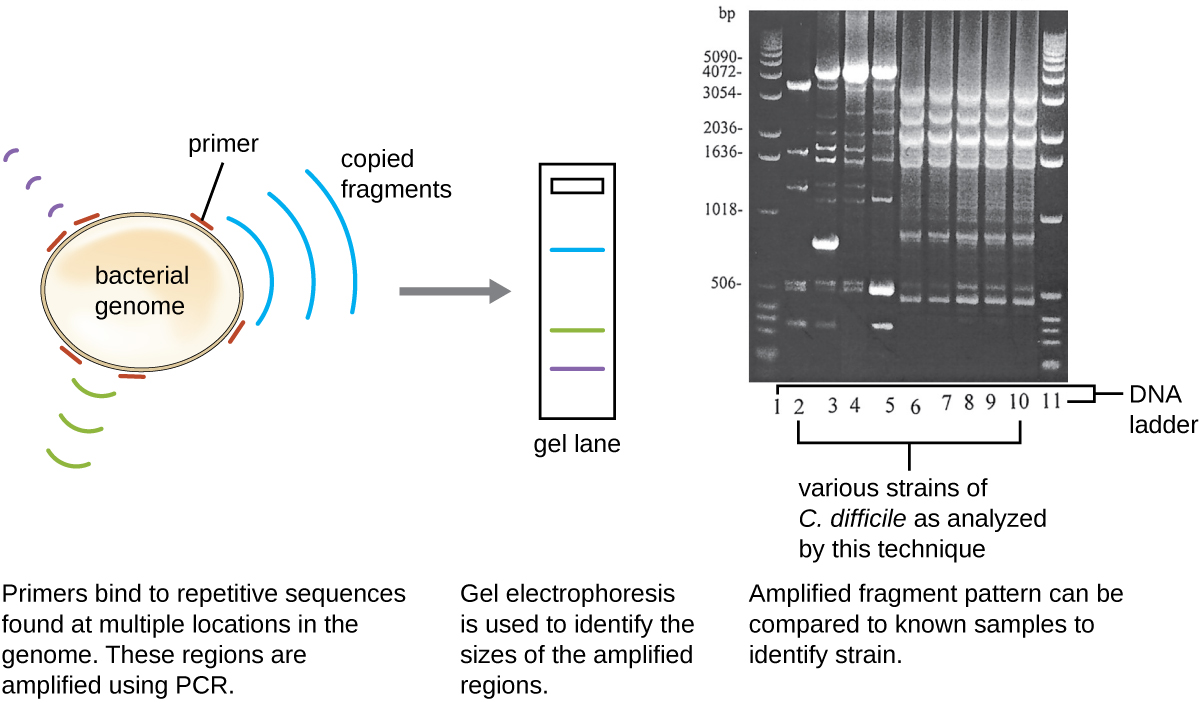
Comme les infections à C. difficile devenaient de plus en plus répandues dans la communauté de Javier, son échantillon a fait l'objet d'une analyse plus approfondie afin de déterminer si la souche spécifique de C. difficile pouvait être identifiée. L'échantillon de selles de Javier a été soumis à un ribotypage et à une analyse PCR répétitive basée sur des séquences (Rep-PCR). Lors du ribotypage, une courte séquence d'ADN entre les gènes de l'ARNr 16S et de l'ARNr 23S est amplifiée et soumise à une digestion par restriction (Figure\(\PageIndex{12}\)). Cette séquence varie selon les souches de C. difficile, de sorte que les enzymes de restriction se coupent à différents endroits. Dans la Rep-PCR, des amorces d'ADN conçues pour se lier à de courtes séquences fréquemment retrouvées dans le génome de C. difficile ont été utilisées pour la PCR. Après digestion par restriction, une électrophorèse sur gel d'agarose a été réalisée dans les deux types d'analyse afin d'examiner les patrons de bandes résultant de chaque procédure (Figure\(\PageIndex{13}\)). La Rep-PCR peut être utilisée pour sous-typer davantage divers ribotypes, augmentant ainsi la résolution pour détecter les différences entre les souches. Le ribotype de la souche infectant Javier s'est avéré être le ribotype 27, une souche connue pour sa virulence accrue, sa résistance aux antibiotiques et sa prévalence accrue aux États-Unis, au Canada, au Japon et en Europe. 1
Exercice\(\PageIndex{5}\)
- En quoi les patrons de bandes diffèrent-ils entre les souches de C. difficile ?
- Pourquoi pensez-vous que les tests de laboratoire n'ont pas permis de détecter directement la production de toxines ?
Exercice\(\PageIndex{6}\)
- En quoi la PCR est-elle similaire au processus naturel de réplication de l'ADN dans les cellules ? En quoi est-ce différent ?
- Comparez la RT-PCR et la qPCR en fonction de leurs objectifs respectifs.
- Dans le séquençage par terminaison de chaîne, comment détermine-t-on l'identité de chaque nucléotide d'une séquence ?
Concepts clés et résumé
- La recherche d'un gène d'intérêt dans un échantillon nécessite l'utilisation d'une sonde d'ADN monocaténaire marquée par une balise moléculaire (généralement de la radioactivité ou de la fluorescence) capable de s'hybrider avec un acide nucléique monocaténaire complémentaire présent dans l'échantillon.
- L'électrophorèse sur gel d'agarose permet de séparer les molécules d'ADN en fonction de leur taille.
- L'analyse du polymorphisme de la longueur des fragments de restriction (RFLP) permet de visualiser par électrophorèse sur gel d'agarose des variants distincts d'une séquence d'ADN provoqués par des différences dans les sites de restriction.
- L'analyse par transfert Southern permet aux chercheurs de trouver une séquence d'ADN particulière dans un échantillon, tandis que l'analyse par transfert nordique permet aux chercheurs de détecter une séquence d'ARNm particulière exprimée dans un échantillon.
- La technologie des microréseaux est une technique d'hybridation des acides nucléiques qui permet d'examiner plusieurs milliers de gènes à la fois afin de détecter des différences dans les gènes ou les modèles d'expression génique entre deux échantillons d'ADN génomique ou d'ADNc,
- L'électrophorèse sur gel de polyacrylamide (PAGE) permet de séparer les protéines selon leur taille, en particulier si les charges protéiques natives sont masquées par un prétraitement au SDS.
- La réaction en chaîne par polymérase permet l'amplification rapide d'une séquence d'ADN spécifique. Les variations de la PCR peuvent être utilisées pour détecter l'expression de l'ARNm (PCR par transcriptase inverse) ou pour quantifier une séquence particulière dans l'échantillon d'origine (PCR en temps réel).
- Bien que le développement du séquençage de l'ADN de Sanger ait été révolutionnaire, les progrès du séquençage de nouvelle génération permettent le séquençage rapide et peu coûteux des génomes de nombreux organismes, accélérant ainsi le volume de nouvelles données de séquence.
Notes
- 1 Patrizia Spigaglia, Fabrizio Barbanti, Anna Maria Dionisi et Paola Mastrantonio. « Isolats de Clostridium difficile résistants aux fluoroquinolones en Italie : émergence du ribotype PCR 018. » Journal de microbiologie clinique 48 n° 8 (2010) : 2892—2896.