
17.3 : Séquençage du génome entier
- Page ID
- 189733

Compétences à développer
- Décrire trois types de séquençage
- Définir le séquençage du génome entier
Bien que des progrès importants aient été réalisés dans les sciences médicales ces dernières années, les médecins sont toujours déconcertés par certaines maladies et ils utilisent le séquençage du génome entier pour aller au fond du problème. Le séquençage du génome entier est un processus qui permet de déterminer la séquence d'ADN d'un génome entier. Le séquençage du génome entier est une approche par force brute pour résoudre des problèmes lorsqu'une base génétique est au cœur d'une maladie. Plusieurs laboratoires fournissent désormais des services de séquençage, d'analyse et d'interprétation de génomes entiers.
Par exemple, le séquençage de l'exome entier est une alternative moins coûteuse au séquençage du génome entier. Lors du séquençage de l'exome, seules les régions codantes de l'ADN productrices d'exons sont séquencées. En 2010, le séquençage de l'exome entier a été utilisé pour sauver un jeune garçon dont les intestins présentaient de multiples abcès mystérieux. L'enfant a subi plusieurs opérations du côlon sans aucun soulagement. Enfin, un séquençage de l'exome entier a été réalisé, qui a révélé un défaut dans une voie contrôlant l'apoptose (mort cellulaire programmée). Une greffe de moelle osseuse a été utilisée pour surmonter cette maladie génétique, permettant ainsi de guérir le garçon. Il a été la première personne à être traitée avec succès sur la base d'un diagnostic posé par séquençage de l'exome entier. Aujourd'hui, le séquençage du génome humain est plus facilement accessible et peut être réalisé en un jour ou deux pour environ 1 000$.
Stratégies utilisées dans le séquençage des projets
La technique de séquençage de base utilisée dans tous les projets de séquençage modernes est la méthode de terminaison de chaîne (également connue sous le nom de méthode didéoxy), développée par Fred Sanger dans les années 1970. La méthode de terminaison de chaîne implique la réplication de l'ADN d'une matrice monocaténaire à l'aide d'une amorce et d'un désoxynucléotide ordinaire (dNTP), qui est un monomère, ou une unité unique, de l'ADN. L'amorce et le dNTP sont mélangés à une petite proportion de didésoxynucléotides marqués par fluorescence (DDNTP). Les DDNTP sont des monomères qui ne possèdent pas de groupe hydroxyle (—OH) au site auquel un autre nucléotide se fixe habituellement pour former une chaîne (Figure\(\PageIndex{1}\)).
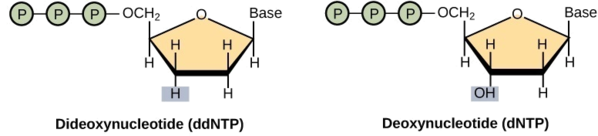
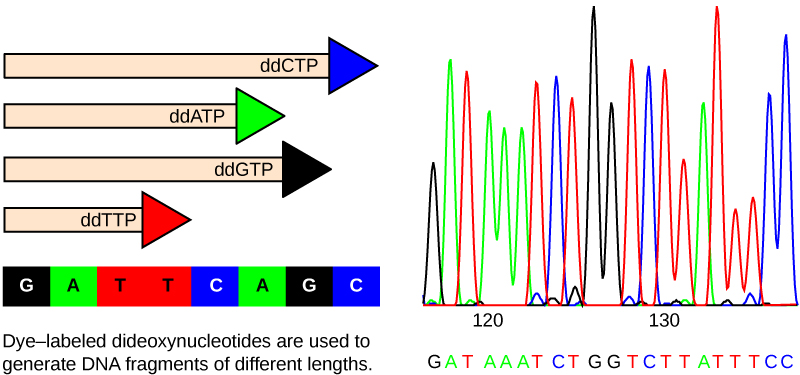
Chaque DDnTP est marqué avec une couleur de fluorophore différente. Chaque fois qu'un ddNTP est incorporé dans le brin complémentaire en croissance, il met fin au processus de réplication de l'ADN, ce qui se traduit par de multiples brins courts d'ADN répliqué qui se terminent chacun à un point différent pendant la réplication. Lorsque le mélange réactionnel est traité par électrophorèse sur gel après avoir été séparé en brins simples, les multiples brins d'ADN nouvellement répliqués forment une échelle en raison de leurs tailles différentes. Comme les DDNTP sont marqués par fluorescence, chaque bande du gel reflète la taille du brin d'ADN et du ddNTP qui a mis fin à la réaction. Les différentes couleurs des DDNTP marqués au fluorophore aident à identifier le DDnTP incorporé à cette position. La lecture du gel sur la base de la couleur de chaque bande de l'échelle produit la séquence du fil modèle (Figure\(\PageIndex{2}\)).
Premières stratégies : séquençage au fusil de chasse et séquençage final par paire
Dans la méthode de séquençage au fusil de chasse, plusieurs copies d'un fragment d'ADN sont découpées au hasard en de nombreux petits morceaux (un peu comme ce qui arrive à une cartouche ronde tirée avec un fusil de chasse). Tous les segments sont ensuite séquencés à l'aide de la méthode de séquençage en chaîne. Ensuite, à l'aide d'un ordinateur, les fragments sont analysés pour voir où leurs séquences se chevauchent. En faisant correspondre les séquences qui se chevauchent à la fin de chaque fragment, la séquence d'ADN complète peut être reformée. Une séquence plus grande qui est assemblée à partir de séquences plus courtes superposées est appelée contig. Par analogie, imaginez que quelqu'un possède quatre copies d'une photographie de paysage que vous n'avez jamais vue auparavant et que vous ne savez rien de la façon dont elle doit apparaître. La personne déchire ensuite chaque photographie avec ses mains, de sorte que des pièces de tailles différentes soient présentes sur chaque copie. La personne mélange ensuite toutes les pièces et vous demande de reconstituer la photographie. Dans l'un des plus petits morceaux, vous voyez une montagne. Dans une pièce plus grande, vous voyez que la même montagne se trouve derrière un lac. Un troisième fragment ne montre que le lac, mais il révèle qu'il y a une cabane au bord du lac. Par conséquent, en regardant les informations qui se chevauchent dans ces trois fragments, vous savez que la photo montre une montagne derrière un lac avec une cabane sur sa rive. C'est le principe qui sous-tend la reconstruction de séquences d'ADN entières à l'aide du séquençage au fusil de chasse.
À l'origine, le séquençage au fusil de chasse n'analysait qu'une extrémité de chaque fragment pour détecter les chevauchements. Cela était suffisant pour le séquençage de petits génomes. Cependant, le désir de séquencer des génomes plus importants, tels que celui d'un être humain, a conduit au développement du séquençage à double canon, plus officiellement connu sous le nom de séquençage par paires. Dans le séquençage par paires, les deux extrémités de chaque fragment sont analysées pour détecter tout chevauchement. Le séquençage par paire est donc plus fastidieux que le séquençage au fusil de chasse, mais il est plus facile de reconstruire la séquence car davantage d'informations sont disponibles.
Séquençage de nouvelle génération
Depuis 2005, les techniques de séquençage automatisé utilisées par les laboratoires s'inscrivent dans le cadre du séquençage de nouvelle génération, qui est un groupe de techniques automatisées utilisées pour le séquençage rapide de l'ADN. Ces séquenceurs automatisés à faible coût peuvent générer des séquences de centaines de milliers ou de millions de fragments courts (25 à 500 paires de bases) en l'espace d'une journée. Ces séquenceurs utilisent des logiciels sophistiqués pour passer à travers le fastidieux processus de mise en ordre de tous les fragments.
Evolution Connection : comparaison de séquences
Un alignement de séquences est un arrangement de protéines, d'ADN ou d'ARN ; il est utilisé pour identifier les régions de similitude entre les types ou les espèces de cellules, ce qui peut indiquer la conservation de fonctions ou de structures. Les alignements de séquences peuvent être utilisés pour construire des arbres phylogénétiques. Le site Web suivant utilise un logiciel appelé BLAST (outil de recherche d'alignement local de base).
Sous « Basic Blast », cliquez sur « Nucleotide Blast ». Entrez la séquence suivante dans le grand champ « séquence de requête » : ATTGCTTCGATTGCA. Sous la case, trouvez le champ « Espèce » et tapez « humain » ou « Homo sapiens ». Cliquez ensuite sur « BLAST » pour comparer la séquence saisie aux séquences connues du génome humain. Il en résulte que cette séquence se trouve à plus de cent endroits du génome humain. Faites défiler vers le bas le graphique avec les barres horizontales et vous verrez une brève description de chacun des résultats correspondants. Choisissez l'un des résultats en haut de la liste et cliquez sur « Graphismes ». Cela vous amènera à une page qui indique où se trouve la séquence dans l'ensemble du génome humain. Vous pouvez déplacer le curseur qui ressemble à un drapeau vert d'avant en arrière pour voir les séquences situées immédiatement autour du gène sélectionné. Vous pouvez ensuite revenir à la séquence sélectionnée en cliquant sur le bouton « ATG ».
Utilisation de séquences du génome entier d'organismes modèles
Le premier génome complètement séquencé était celui d'un virus bactérien, le bactériophage fx174 (5368 paires de bases) ; Fred Sanger l'a fait à l'aide du séquençage au fusil de chasse. Plusieurs autres génomes d'organites et de virus ont ensuite été séquencés. Le premier organisme dont le génome a été séquencé était la bactérie Haemophilus influenzae ; Craig Venter l'a fait dans les années 1980. Environ 74 laboratoires différents ont collaboré au séquençage du génome de la levure Saccharomyces cerevisiae, qui a débuté en 1989 et s'est achevé en 1996, car il était 60 fois plus gros que tout autre génome séquencé. En 1997, les séquences génomiques de deux organismes modèles importants étaient disponibles : la bactérie Escherichia coli K12 et la levure Saccharomyces cerevisiae. Les génomes d'autres organismes modèles, tels que la souris Mus musculus, la mouche des fruits Drosophila melanogaster, le nématode Caenorhabditis elegans et les humains Homo sapiens sont maintenant connus. De nombreuses recherches fondamentales sont effectuées sur les organismes modèles, car les informations peuvent être appliquées à des organismes génétiquement similaires. Un organisme modèle est une espèce étudiée comme modèle afin de comprendre les processus biologiques d'autres espèces représentées par l'organisme modèle. Le séquençage de génomes entiers contribue aux efforts de recherche sur ces organismes modèles. Le processus qui consiste à associer des informations biologiques à des séquences de gènes s'appelle l'annotation du génome. L'annotation des séquences de gènes facilite les expériences de base en biologie moléculaire, telles que la conception d'amorces de PCR et de cibles d'ARN.

Utilisations des séquences génomiques
Les puces à ADN sont des méthodes utilisées pour détecter l'expression des gènes en analysant un ensemble de fragments d'ADN fixés sur une lame de verre ou une puce de silicium afin d'identifier des gènes actifs et d'identifier des séquences. Près d'un million d'anomalies génotypiques peuvent être découvertes à l'aide de puces, tandis que le séquençage du génome entier peut fournir des informations sur les six milliards de paires de bases du génome humain. Bien que l'étude des applications médicales du séquençage du génome soit intéressante, cette discipline tend à s'attarder sur le fonctionnement anormal des gènes. La connaissance de l'ensemble du génome permettra de découvrir rapidement les maladies futures et d'autres troubles génétiques, ce qui permettra de prendre des décisions plus éclairées concernant le mode de vie, les médicaments et la naissance d'enfants. La génomique en est encore à ses balbutiements, mais il pourrait devenir courant un jour d'utiliser le séquençage du génome entier pour dépister chaque nouveau-né afin de détecter des anomalies génétiques.
Outre les maladies et les médicaments, la génomique peut contribuer au développement de nouvelles enzymes qui convertissent la biomasse en biocarburant, ce qui se traduit par une augmentation de la production de cultures et de carburants et une baisse des coûts pour le consommateur. Ces connaissances devraient permettre de meilleures méthodes de contrôle des microbes utilisés dans la production de biocarburants. La génomique pourrait également améliorer les méthodes utilisées pour surveiller l'impact des polluants sur les écosystèmes et aider à éliminer les contaminants environnementaux. La génomique a permis le développement de produits agrochimiques et pharmaceutiques qui pourraient bénéficier à la science médicale et à l'agriculture.
Cela semble formidable de posséder toutes les connaissances que nous pouvons tirer du séquençage du génome entier ; cependant, les humains ont la responsabilité d'utiliser ces connaissances à bon escient. Dans le cas contraire, il pourrait être facile d'abuser du pouvoir de ces connaissances, ce qui conduirait à une discrimination fondée sur la génétique d'une personne, le génie génétique humain et d'autres préoccupations éthiques. Ces informations peuvent également entraîner des problèmes juridiques concernant la santé et la confidentialité.
Résumé
Le séquençage du génome entier est la dernière ressource disponible pour traiter les maladies génétiques. Certains médecins utilisent le séquençage du génome entier pour sauver des vies. La génomique a de nombreuses applications industrielles, notamment le développement de biocarburants, l'agriculture, les produits pharmaceutiques et le contrôle de la pollution. Le principe de base de toutes les stratégies de séquençage modernes implique la méthode de séquençage par terminaison de chaîne.
Bien que les séquences du génome humain fournissent des informations essentielles aux professionnels de la santé, les chercheurs utilisent des séquences du génome entier d'organismes modèles pour mieux comprendre le génome de l'espèce. L'automatisation et la diminution du coût du séquençage du génome entier pourraient conduire à une médecine personnalisée à l'avenir.
Lexique
- méthode d'arrêt de chaîne
- méthode de séquençage de l'ADN utilisant des didésoxynucléotides marqués pour mettre fin à la réplication de l'ADN ; elle est également appelée méthode didésoxy ou méthode de Sanger
- contig
- séquence d'ADN plus grande assemblée à partir de séquences plus courtes superposées
- désoxynucléotide
- monomère individuel (unité unique) d'ADN
- didésoxynucléotide
- monomère individuel d'ADN dépourvu d'un groupe hydroxyle (—OH)
- Microarray à ADN
- méthode utilisée pour détecter l'expression génique en analysant un ensemble de fragments d'ADN fixés sur une lame de verre ou une puce de silicium afin d'identifier des gènes actifs et d'identifier des séquences
- annotation du génome
- processus d'attachement d'informations biologiques à des séquences de gènes
- organisme modèle
- espèce étudiée et utilisée comme modèle pour comprendre les processus biologiques chez d'autres espèces représentées par l'organisme modèle
- séquençage de nouvelle génération
- groupe de techniques automatisées utilisées pour le séquençage rapide de l'ADN
- séquençage des fusils de chasse
- méthode utilisée pour séquencer plusieurs fragments d'ADN afin de générer la séquence d'un gros morceau d'ADN
- séquençage du génome entier
- processus qui détermine la séquence d'ADN d'un génome entier